Patient Bone Marrow Aspiration to Explore the Cyclooxygenases (COXs) Involvement in Multiple Myeloma
Received Date: December 16, 2020 Accepted Date: January 16, 2021 Published Date: January 18, 2021
doi: 10.17303/jcrto.2021.9.103
Citation: Scilimati A (2021) Patient Bone Marrow Aspiration to Explore the Cyclooxygenases (COXs) Involvement in Multiple Myeloma. J Cancer Res Therap Oncol 9: 1-19.
Abstract
Limited data of Cyclooxygenases (COXs) role in Multiple Myeloma (MM) onset and/or progression are available. Then, seven human MM cell lines (hMMCLs), representative of different disease stages, were tested to evaluate the COXs expression level. Cell viability upon treatments with COX inhibitors alone or in combination with clinically used anti-MM drugs was also determined. All tested drugs exerted a fair antiproliferative activity on hMMCLs and did not induce changes of the two isoenzymes expression extent. Biosynthesis of COXs-mediated PGE2 and TXB2 and NF-κB activation were measured after cell treatment with COX inhibitors and in combination with the anti-MM drugs. No marked synergistic cytotoxic effect was observed by their coadministration. Measuring the PGE2 and TXB2 formed amount, it seems that the used drugs exert their action by a COXs independent mechanism, at least in the chosen hMMCLs and then also in bone marrow stromal cells (BMSCs), derived from MM patient bone marrow aspirates, as an attempt to build the in vivo disease condition. Our data confirmed the permissive microenvironment (BMSCs) role in MM cell proliferation.
Keywords: Multiple Myeloma; Multiple Myeloma Patient Bone Marrow Aspirates; Cyclooxygenase (COX); 2D Cell Culture; Mofezolac; Anti-Multiple Myeloma Drugs
Abbreviations: AA: Achidonic Acid; ASA: Aspirin; BMSCs: Bone Marrow Stromal Cells; BRZ: Bortezomib; CCK-8: Cell Counting kit-8; CEL: Celecoxib; COX: Cyclooxygenase; DEX: Dexamethasone; DMSO: Dimethyl Sulfoxide; LPS: Lipopolysaccharide; MDCK: Madin-Darby Canine Kidney cells; MM: Multiple Myeloma; MOF: Mofezolac; PBS: Phosphate-Buffered Saline; PFA: Paraformaldehyde; PG: Prostaglandin; SC: SC 560; THA: Thalidomide
Introduction
Multiple Myeloma (MM) is still an incurable malignant disease affecting plasma cells. It is the second most common hematologic malignancy and is characterized by a marked genetic heterogeneity [1]. Tumor cells are mainly located in the bone marrow, where their phenotype features (survival, apoptosis, homeostasis, proliferation, invasion and angiogenesis) are affected by the interactions with stromal cells, hematopoietic cells, and extracellular matrix. Myeloma cells originate from follicle center B-lymphocytes by somatic hypermutation of antigen binding sites and immunoglobulin isotypes switching to differentiate to plasma cells. This suggests a critical role for antigenic and inflammatory stimulation in myelomagenesis. Disease-specific therapy on symptomatic MM patients eligible for intensive treatment includes an induction treatment with high-dose dexameth asone (DEX) and at least one “new agent”, i.e. the proteasome inhibitor bortezomib (BRZ), and the immunomodulatory drugs thalidomide (THA) or lenalidomide (LEN) (Figure 1). Such an induction treatment, acting on tumor plasma cells as well as on tumor milieu, decreases the tumor mass and improves patient status.
DEX (Figure 1) is cytotoxic towards tumor cells and inhibits the production of pro- inflammatory mediators that support tumor cell growth [2]. BRZ inhibits protein degradation through the proteasome, resulting in tumor cell cycle blockade and apoptosis [3]. LEN and THA have pleiotropic actions not yet fully elucidated, such as immune cell stimulation, angiogenesis suppression and cell-cycle inhibition and apoptosis induction [4].
65-70 Years old patient’s ineligible for intensive medicaments are treated with melphalan and prednisone, in combination with THA or BRZ and eventually LEN [5].
The widespread use of high-dosage chemotherapy with hematopoietic stem cell transplantation, and the introduction of the new agents BRZ, THA and LEN demonstrated that prognosis of advanced disease can be significantly improved [6]. However, a complete and lasting recovery from the disease is not achievable, and the patients invariably relapse and become resistant to all treatment options (or cannot be further treated because of adverse side effects onset).
The relapsed/resistant phenotype can be attributed to residual treatment-resistant tumor cells, located in focal lesions and responsible for disease relapse. A real cure should completely eradicate these tumor cells, and to this end, studies aimed at understanding the mechanisms driving drug sensitivity/resistance should be pursued, and tools for modulating such mechanisms should be investigated.
In this context, the P-glycoprotein (P-gp) role should also be considered in stating the overall effectiveness of MM treatment. Multidrug resistance (MDR) mediated by P-gp is one of the major reasons for the failure of most cancer treatments. It is proven that non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, curcumin, ibuprofen and NS-398 downregulate P-gp expression [7], thus, overcoming P-gp-mediated drug resistance. These results provide the rationale to study NSAIDs as P-gp function modulators in MM. Cyclooxygenase is the main NSAIDs target [8]. Two isoforms of COX are known, as product of different genes, COX-1, and COX-2 [9,10]. Cyclooxygenase (COX) is the enzyme in part responsible for the biosynthesis of prostaglandins (PGs), catalyzing the conversion of arachidonic acid (AA) into PGH2, the precursor of PGs. AA, in turn, is released from the cell membrane upon inflammatory and mitogen stimuli. PGs, particularly PGE2, are mediators of inflammation and angiogenesis, and support the growth of several solid tumors [11,12]. PGE2 also stimulates gene transcription, influences mitogenesis of normal human bone cells, and promotes tumor metastasis. Both known COX isoenzymes contribute to PGE2 production, even though COX-1 in normal tissue seems to be its major source [13].
Instead, in several human cancer types, COX-2 expression actively supports tumor cell growth and is considered as an indicator of poor prognosis. Accordingly, COX-2 is a useful target for chemo-preventive and therapeutic intervention for solid and, recently, also hematological malignancies [14-17]. A few studies have examined COX-2 in chronic myelogenous leukemia and non-Hodgkin lymphoma, and very limited data are available in MM [18-22]. COX-1 involvement in tumorigenesis was generally less investigated than COX-2. However, in some type of tumors, such as the ovarian cancer [23-25], COX-1 is the major PGE2 source, in turn associated with the induction of angiogenic factors like vascular endothelial growth factor, basic fibroblast growth factor, angiopoietin-1. Hence, it is relevant to control PGE2 biosynthesis by inhibiting specifically one or both COXs.
Several studies showed an improved cytotoxicity when anti-MM drugs (i.e., DEX, BRZ, LEN and THA) are used together with COX-2 inhibitors (i.e., celecoxib). Recently, positive effects of COX-1 inhibitors on cytotoxicity, cell cycle and apoptosis in two different MM cell lines (RPMI-8226 and NCI-H929) with a different COX expression profile were reported [26, 27]. Overall, the distinct role of each COX isoform in MM is still unclear, also because very few data are available on the effect of COX-1 inhibition.
Based on the best of our knowledge, only Ding et al. investigated the expression of both COX isoforms and found a variable expression in several myeloma cell lines [28]. As a consequence, it is valuable any investigation aimed at both clarifying COXs role in MM and the usefulness of their inhibitors, as potential chemotherapeutic agents, to be used alone or in combination with the currently prescribed anti-multiple myeloma drugs.
In this regard, known COX inhibitors (Figure 2) with different selectivity towards the two isoforms [SC 560 (SC) and mofezolac (MOF), as selective COX-1 inhibitors; celecoxib (CEL) as a selective COX-2 inhibitor; and aspirin (ASA) as dual COX inhibitor] were investigated alone or in combination with some clinically used anti-MM drugs (Figure 1) on a panel of human MM cell lines (hMMCLs), selected to be representative of the molecular heterogeneity of MM [22, 26-27].
The effect of those treatments on cell growth, COXs expression and P-gp modulation was evaluated. PGE2 and thromboxane (TXB2) formation, and NF-kB activation were also determined as a measure of COXs activity.
Tumor cells, primarily located in the bone marrow milieu, interact with stromal cells, hematopoietic cells, and extracellular matrix affecting the most important aspects of the malignant cell phenotype (i.e. survival/apoptosis, homeostasis, proliferation, invasion and angiogenesis). Particularly, bone marrow stromal cells (BMSCs) create a permissive microenvironment for MM cell proliferation, angiogenesis, metastasis, and development of drug resistance [29]. It was hypothesized that, the pro-tumoral effect of BMSCs would correlate, at least in part, with COX activation since pro- inflammatory mediators were found to be up regulated in it [30]. Thus, a strategy to reduce BMSCs tumor permissive effect could be through COX inhibition.
Herein, we report the results of the co-administration of COXs inhibitors with anti-MM drugs in seven human MM cell lines, chosen to be representative of various MM stages [31, 32], and to human bone marrow stromal cells derived from bone marrow aspirations of patients with active MM disease with the aim to mime in vivo tumor milieu condition.
Materials and Methods
Materials
Cell culture reagents were purchased from EuroClone (Milan, Italy). SC 560, celecoxib, aspirin, RIPA buffer, protease inhibitor cocktail, Calcein-AM, bisBenzimide H 33342 trihydrochloride and Cell Counting kit (CCK-8) were obtained from Sigma-Aldrich (Milan, Italy). Mofezolac was prepared in our laboratory [33]. Anti-β-actin (Prod #AM4302; Lot 00495954), anti-COX-1 (COX- 111; Prod # 35-8100; Lot QD215497) and anti-COX-2 (COX-229; Prod # 35-8200; Lot RE231769) were purchased from Thermo Fisher Scientific Italia (Monza, Italy) antibodies. Anti-mouse secondary peroxidase antibody (Cat. #170-6516; Lot L005680 A) and all reagents for Western Blotting were purchased from Bio-Rad Laboratories Srl (Milan, Italy).
Human multiple myeloma cell lines (hMMCLs) are widely used for their representation of primary myeloma cells and to study the biology of MM. For this purpose, seven hMMCLs delineated by Arkans high glucose Cancer Research Center (ACRC) classification were chosen for the studies herein described: human NCI-H929, KMS-12-BM and SK-MM-2 cells were obtained from the Leibniz Institute DSMZ (Germany); human RPMI-8226, U266-B1, MM1R, and MM1S MM cells were purchased from ATCC (Manassas, VA).
hNCI-H929 was established from the pleural effusion of a 62-year-old white woman with myeloma (IgAkappa) at relapse; hKMS-12-BM was established from the bone marrow of a 64-year-old woman with MM and hSK-MM-2 was established from the peripheral blood of a 54-year-old man with plasma cell leukemia (Igkappa) (refractory, terminal state); plasma cell leukemia is related to MM. hRPMI-8226 was established from the peripheral blood of a 61-year-old man with MM (IgG lambda-type) at diagnosis, hU266-B1 was established from the peripheral blood of a 53-year-old man with IgE-secreting myeloma (refractory, terminal state), MM1R, and MM1S MM cells were independently created from the parental cell line MM-1 to represent models of resistance and sensitivity, respectively, to DEX.
NCI-H929 cells were grown in RPMI-1640 supplemented with 20% fetal bovine serum, 50 μM β- mercaptoethanol, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified incubator at 37 °C and 5% CO2 atmosphere. KMS-12-BM and SK- MM-2 cells were grown in RPMI-1640 supplemented with 20% fetal bovine serum, 2 mM L- glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified incubator at 37 °C and 5% CO2 atmosphere. MM1R and MM1S cells were grown in RPMI-1640 supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, 1.4 M glucose, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified incubator at 37 °C and 5% CO2 atmosphere.
RPMI-8226 and U266 B1 cells were grown in RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified incubator at 37 °C and 5% CO2 atmosphere.
HEK-293-derived cell lines stably and inducibly expressing hCOX-IRES-mPGES-1 were generated in tetracycline-inducible mammalian expression system [34]. HEK-293 COX-1 and HEK-293 COX-2 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% no-heat-inactivated fetal bovine serum, supplemented with 2 mM L- glutamine, 100 μg/ml hygromycin B, and 6.5 μg/ml blasticidin S, in a humidified incubator at 37 °C and 5% CO2 atmosphere.
MDCK-MDR1, MDCK-MRP1 and MDCK-BCRP cells are a gift of Prof. P. Borst (NKI-AVL Institute, Amsterdam). Caco-2 and MDCK cells were grown in DMEM high glucose supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, in a humidified incubator at 37 °C and 5% CO2 atmosphere. Each cell line was used from passage 5 to passage 20.
Bone marrow mononuclear cells derived from MM patient bone marrow aspiration
Bone marrow samples were obtained from 6 patients with symptomatic MM according to the International Myeloma Working Group criteria, in turn used to classify monoclonal gammopathies, multiple myeloma and related disorders [35]. The study protocol was approved by the Medical School Ethics Committee of the University of Bari and conformed to the good clinical practice guidelines of the Italian Ministry of Health. Written informed consent was obtained from each subject in accordance with the Declaration of Helsinki.
Bone marrow mononuclear cells (BMMCs) were isolated by Ficoll-Paque Plus (GE Healthcare Life Sciences) density gradient centrifugation. BMSCs were obtained from adherent BMMCs cultured for 4 weeks [36]. Specifically, BMMCs were left to adhere in culture dishes (BD Falcon) in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin disposables and reagent purchased from Euroclone. After 1 week, suspension cells were removed, and adherent cells were expanded. After 3 weeks, stromal cells were harvested.
BMSCs were cultured in DMEM high glucose medium (Euroclone) supplemented with 20% heat- inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin in a humidified incubator at 37 °C in 5% CO2 and 95% relative humidity.
hMMCLs viability determination
Determination of cell growth was performed using the CCK-8 assay at 48 hours. On day 1, 50,000 cells/well were seeded into 96-well plates in a volume of 50 μL. On day 2, the various drug concentrations (cell viability paragraph) were added in a volume of 50 μL. In all the experiments, the various solvents (EtOH, DMSO) used to solubilize the drugs were added in each control to evaluate a possible solvent cytotoxic effect. At the established incubation time (48 hours) in the presence of the drugs, CCK-8 (10 μL) was added to each well, and after 3-4 hours incubation at 37 °C, the absorbance values at ⅄ = 450 nm were read on the microplate reader Victor 3 (PerkinElmer Life Sciences).
Western blotting analysis
All cells were washed twice with 10 ml phosphate buffered saline (PBS), scraped in 1 ml PBS and centrifuged for 10 min at 1,500 rpm, at 4 °C. Proteins were extracted from cells by homogenization in cold RIPA buffer (Sigma-Aldrich) containing 1X protease inhibitor cocktail and centrifuged for 10 min at 14,000 rpm, at 4 °C. The supernatant was recovered, and the protein concentration was measured using DC Protein assay Reagent Kit (BIO-RAD). 30 μg of protein extract was separated on 10% polyacrylamide gel (BIO-RAD) and then transferred onto a polyvinylidene difluoride membrane (PVDF) by Trans-Blot Turbo Transfer System (BIO-RAD). Membrane was blocked for 1 hour at room temperature (r.t.) with blocking buffer (1% Blotting Grade Blocker, 0.1% Tween 20 in Tris-buffered saline, TBS). The membrane was then incubated with either anti-COX-1 (1:500 mouse monoclonal, overnight at 4 °C), anti-COX-2 (1:500, mouse monoclonal, overnight at 4 °C) or anti-β-actin (1:1000 mouse monoclonal, 1 hour at room temperature) antibodies, diluted in blocking buffer. After incubation time, the membrane was washed with washing buffer (0.1% Tween-20 in TBS) three times and incubated with a secondary peroxidase antibody (1:3000 anti- mouse) for 1 hour at r.t. After washing, the membrane was treated with the enhanced chemiluminescence (ECL, BIO-RAD) according to the manufacturer’s instructions and the blot was visualized by UVITEC Cambridge (Life Technologies).
The expression level was evaluated by densitometric analysis using UVITEC Cambridge software (Life Technologies), and β-actin expression level was used as loading control to normalize the sample values.
Cells were incubated for 24 hours with (+) or without (-) LPS at different concentrations (10 μg/ml, 50 μg/ml or 100 μg/ml) and the cell homogenates (30 μg protein) were applied to Western blotting. HEK-293 COX-1 and HEK-293 COX-2 were used as positive controls for COX-1 and COX-2 expression, respectively [34]. The protein level of β-actin was used as loading control. Expression of COX-1 and COX-2 proteins in NCI-H929 and SK-MM-2 cell lines upon treatment with mofezolac alone or in combination with DEX or THA was determined. Cells were incubated for 48 hours with (+) or without (-) mofezolac (75 μM in NCI-H929 and 80 μM in SK- MM-2), DEX (35 μM in NCI-H929 and 80 nM in SK-MM-2) or THA (45 μM in NCI-H929 and 65 μM in SK-MM-2) used at different concentrations on the basis of the respective EC50 values. The cell homogenates (30 μg protein) were applied to Western blotting. The protein level of β-actin was used as loading control.
PGE2 and TXB2 assay
NCI-H929 cells were cultured in 6-well plates at a density of 1×106 cells/well. Then, the cells were treated with BRZ, DEX and THA alone or in combination with mofezolac or celecoxib and maintained at 37 °C for 48 hours in a humidified air containing a 5% CO2. PGE2 levels were determined in the supernatant using competitive binding immunoassay (Item no. 514010, Cayman Chemical, Ann Arbor, MI, USA) following the manufacturer’s instructions. TXB2 levels were determined in the supernatant using a competitive binding immunoassay (Item no. 10004023, Cayman Chemical, Ann Arbor, MI, USA) following the manufacturer’s instructions [37]. Untreated cells were included as a control. The optical density was measured at λ = 412 nm with precision microplate reader and the amount of PGE2 (ng/mL) was calculated using a PGE2 standard curve.
NF-κB activation assay
NCI-H929 cells were cultured in 6-well plates at a density of 1×106 cells/well. Then, the cells were treated with (+) or without (-) LPS and in a separate experiment with BRZ, DEX and THA alone or in combination with mofezolac or celecoxib and maintained at 37 °C for 48 hours in a humidified air containing 5% CO2. Nuclear proteins were isolated by using a compartmental protein extraction kit (Item no. 10009277, Cayman Chemical, Ann Arbor, MI, USA) following the manufacturer’s protocol. NF-κB activation was assayed using an ELISA-based NF- κB (p65) Transcription Factor Assay kit (Item no. 10007889, Cayman Chemical, Ann Arbor, MI, USA) containing a 96-well plate with immobilized oligonucleotides encoding an NF-κB consensus site according to the manufacturer’s protocol. The active form of NF-κB contained in the nuclear extract specifically binds to this oligonucleotide. The primary antibody used to detect NF-κB recognizes an epitope on p65 that is accessible only when NF-κB is activated and bound to its target DNA. A horseradish peroxidase (HRP)-conjugated secondary antibody provides a sensitive colorimetric readout that is quantified by a precision microplate reader at λ = 450 nm with a reference wavelength of λ = 655 nm. Untreated cells were included as a control. The amount of the activated NF-κB (ng/mL) was calculated using a NF-κB standard curve.
Calcein-AM experiments
These experiments were carried out as previously described [38-40] with minor modifications. Briefly, each cell line (30,000 cells per well) was seeded into black CulturePlate 96-well plate with 100 μL medium and allowed to become confluent overnight. 100 μL of test compounds were solubilized in the culture medium and added to the monolayers, to the final concentrations ranging from 0.1 to 100 μM.
96-Well plate was incubated at 37 °C for 30 min. Calcein-AM was added in 100 μL of phosphate buffered saline (PBS) to yield a final concentration of 2.5 μM. The plate was incubated for 30 min. Each well was washed 3 times with ice cold PBS. Saline buffer was added to each well and the plate was read with Victor 3 (PerkinElmer) at excitation and emission wavelengths of λ = 485 and 535 nm, respectively. In these experimental conditions, Calcein cell accumulation in the absence and in the presence of tested drugs was evaluated and fluorescence basal level was estimated with untreated cells. In treated wells, the increase of fluorescence with respect to basal level was measured. EC50 values were determined by fitting the fluorescence increase percentage versus log[dose].
Hoechst 33342 fluorescence experiments
These experiments were carried out as already described [38-40] with minor modifications. Briefly, each cell line (30,000 cells per well) was seeded into 96-well black CulturePlate with 100 μL medium and allowed to become confluent overnight. 100 μL of tested drugs were solubilized in culture medium and added to monolayers, with final concentrations ranging from 0.1 to 100 μM. 96-Well plate was incubated at 37 °C for 30 min. Hoechst 33342 was added in 100 μl of phosphate buffered saline (PBS) to reach the final concentration of 8 μM and plate was incubated for 30 min. The supernatants were drained, and the cells were fixed for 20 min under light protection using 100 μL per well of a 4% paraformaldehyde (PFA) solution. Each well was washed 3 times with ice cold PBS. Saline buffer was added to each well and the plate was read with Victor 3 (PerkinElmer) at excitation and emission wavelengths of λ = 340 nm and 485 nm, respectively. In these experimental conditions, Hoechst 33342 accumulation in the absence and in the presence of tested drugs was evaluated and fluorescence basal level was estimated with untreated cells. In treated wells, the increase of fluorescence with respect to basal level was measured. EC50 values were determined by fitting the fluorescence increase percentage versus log[dose].
ATPlite assay
The MDCK-MDR1 cells were seeded into 96-well microplate in 100 μL of complete medium at a density 2×104 cells/well [38-40]. The plate was incubated overnight (O/N) in a humidified atmosphere 5% CO2 at 37 °C. The medium was removed and 100 μL of complete medium either alone or containing different concentrations of tested drugs was added. The plate was incubated for 2 hours in a humidified 5% CO2 atmosphere at 37 °C. 50 μL of mammalian cell lysis solution was added to all wells and the plate was shaken for 5 minutes in an orbital shaker. 50 μL of drug solution was added to all wells and the plate shaken for 5 minutes in an orbital shaker. The plate was dark adapted for ten minutes and the luminescence was measured.
Permeability Experiments
Preparation of Caco-2 monolayer: Caco-2 cells were seeded onto a Millicell® assay system (Millipore), where a cell monolayer is set in between a filter cell and a receiver plate, at a density of 10,000 cells/well [41]. The culture medium was replaced every 48 hours and the cells kept for 21 days in culture. The Trans Epithelial Electrical Resistance (TEER) of the monolayers was measured daily, before and after the experiment, using an epithelial voltohmmeter (Millicell®-ERS).
Generally, TEER values greater than 1000 Ω for a 21 days culture, are considered optimal.
Drug transport experiment: After 21 days of Caco-2 cell growth, the medium was removed from filter wells and from the receiver plate, which were filled with fresh HBSS buffer (Invitrogen). This procedure was repeated twice, and the plates were incubated at 37 °C for 30 min. After this incubation time, the HBSS buffer was removed and drug solutions and reference compounds were added to the filter well at the concentration of 100 μM, while fresh HBSS was added to the receiver plate. The plates were incubated at 37 °C for 2 hours. Afterwards, samples were removed from the apical (filter well) and basolateral (receiver plate) side of the monolayer to measure the permeability.
The apparent permeability (Papp nm/second), was calculated using the following equation:
Papp = [VA/(area × time)] × ([drug]acceptor /[drug]initial)
where VA is the volume (mL) in the acceptor well, area is the surface area of the membrane (0.11 cm2 of the well), time is the total transport time in seconds (7200 sec), [drug]acceptor is the concentration of the drug measured by UV spectroscopy [drug]initial is the initial drug concentration (1×10−4 M) in the apical or basolateral well.
Results and Discussion
COXs expression extent in multiple myeloma cell lines
Arachidonic acid (AA) cascade is involved in several human physio-pathological conditions. It is mediated also by COXs. PGE2, one of the prostaglandins formed in the COX-catalyzed AA transformation, is responsible for gastric mucosa integrity and inducer of some severe adverse events. For this reason, COXs activity and expression extent are worthy to be determined also in this context. COXs expression levels were measured by Western blotting analysis in seven human multiple myeloma cell lines (hMMCLs) [26], such as RPMI-8226, U266-B1 NCI-H929, SK-MM-2, KMS-12-BM, MM1S and MM1R (Figure 3).
As a positive control of COX-1 and COX-2, the engineered cell lines HEK-293 COX-1 and HEK-293 COX-2 [34], that stimulated with 10 μg/mL tetracycline express COX-1 or COX-2, respectively, were used. COX-1 isoenzyme was detected in NCI-H929 and SK-MM-2 cells cultured both in the presence or absence of lipopolysaccharide (LPS). A faint band of COX-1 was seen in the LPS treated MM1R and KMS-12-BM cells. No COX-1 expression was detected in the other cell lines even if they were treated with three different LPS concentrations (10, 50, 100 μg/ml) for 24 hours. When COX-2 isoenzyme was detected with anti-COX-2 monoclonal antibody, the band was only observed in LPS-treated U266-B1 cells, and no protein was seen in the other LPS-treated and non- treated cell lines.
COXs expression level upon different treatments
To investigate if COX-1 expression in NCI-H929 and SK-MM-2, the only two cell lines expressing COX-1, could be affected by the presence of its selective inhibitor and/or currently prescribed anti- multiple myeloma drugs, dexamethasone (DEX) and thalidomide (THA) alone or in combination with mofezolac (MOF) were administered. A faint band of COX-1 protein, with respect to the positive control HEK-293 COX-1 to which the value = 1 was assigned (Table 1), was detected in the untreated NCI-H929 and SK- MM-2.
Untreated NCI-H929 expresses 6% of COX-1 (Table 1), and when cells are co-treated with mofezolac and THA the value reached 17%. Much higher is the increase in SK-MM-2, in which COX-1 expression was 60% upon treatment with DEX alone or 50% in the presence of THA and mofezolac (respect to 3% in the untreated cells). Incubation with THA alone or DEX with mofezolac determined a slight change in COX-1 expression (25 and 28%, respectively). A slight synergistic effect seems due to co-administered mofezolac and THA to SK-MM-2 cell line because a little increase of COX-1 expression is observed.
No COX-2 expression was detected in NCI-H929 and SK-MM-2 treated cell lines.
As above reported (Figure 3) U266-B1 and RPMI-8226 cell lines in absence of any treatment do not express both COX-1 and COX-2. After treatment of the same cell lines with mofezolac alone or in combination with BRZ, DEX or THA again neither COX-1 or COX-2 were detected (Figure S1).
Cell viability
We have, then, examined if cell cytotoxicity exerted by the anti-MM drugs could be affected by the presence of COX inhibitors. In view of the different expression levels of COX-1 and COX-2 in the seven used hMMCLs, we expected to find a different sensitivity of the tumor cell lines to COX inhibitors and eventually to their combinations with anti-MM drugs. The drugs treatment effect was evaluated by CCK-8 assay on cell growth after 48 hours incubation time. COX inhibitors and anti- MM drugs were used at different concentrations, chosen based on their EC50 values previously determined in the same assay conditions.
In NCI-H929 cells, BRZ and DEX were not effective at the concentrations of 35 μM and 2 nM, respectively, whereas THA and lenalidomide determined a moderate cytotoxic effect (~30 %) at their EC50 = 45 μM. In NCI-H929 cell line the antiproliferative activity of BRZ and DEX was slightly improved if combined with SC 560, while the cytotoxic effect registered after performing the combined treatment with the other COX inhibitors was similar to that one obtained after incubation with aspirin, celecoxib and mofezolac alone (Figure S2). Antiproliferative activity of COX inhibitors was higher in combination with THA and lenalidomide, excepted for SC 560 combined with lenalidomide.
SK-MM-2 treatment with 80 μM lenalidomide alone or in combination with 80 μM mofezolac, 80 μM SC 560 or 70 μM celecoxib induced the same cytotoxic effect (31-36%), whereas its combination with 40 μM aspirin led to a drop in cytotoxicity (only 5%), that in turn it is similar to aspirin alone (2%) (Figure S3). The antiproliferative effect remained the same when 65 μM THA and 4 nM BRZ were administered alone or in combination with aspirin and celecoxib, whereas the cytotoxic effect was higher after the combined treatment with mofezolac and SC 560. Aspirin and SC 560 had the same cytotoxicity if administered alone or in combination with 80 nM DEX, whereas a higher cytotoxicity value was observed for the co-administration of DEX and celecoxib, cytotoxicity passes from 5% for DEX alone to 17% for the combination of the two drugs.
In MM1R, BRZ and DEX alone or in combination with COX inhibitors displayed low cytotoxic activity (Figure S4). Lenalidomide and THA treatment showed an improved antiproliferative effect when they were co-adminstered with the three COX-1 inhibitors, aspirin, mofezolac and SC 560. The higher cytotoxic effect observed in the DEX resistant cell line MM1R, upon aspirin treatment could be due to its P-gp downregulating properties [7].
Celecoxib showed the same or lower effect alone or in combination with THA, lenalidomide or DEX.
In RPMI-8226, that does not express COXs, anti-MM drugs cytotoxicity is always increased in the presence of COX inhibitors (Figure S5). This cytotoxicity increasing in a cell line that does not express both cyclooxygenase isoenzymes, leads us to hypothesize that a COX-independent mechanism might be involved.
On the other hand, BRZ showed a strong cytotoxic activity when it was administered alone or in combination with COX inhibitors in KMS-12-BM cell line (Figure S6) another cell line which does not express COXs, at least in these experimental conditions. Treatment with aspirin, celecoxib, or SC 560 alone or in combination with THA, lenalidomide or DEX displayed very small changes in antiproliferative activity, while a higher cytotoxicity was registered when they were used in combination with mofezolac.
In U266-B1 cells, only SC 560 displayed a higher cytotoxicity when it was combined with anti-MM drugs, whereas the other COX inhibitors showed the same antiproliferative activity alone or in a combined treatment (Figure S7).
MM1S cell line were the less sensitive to the various treatments. Only BRZ, combined with SC 560 or celecoxib was found to have a good cytotoxic effect (Figure S8).
To evaluate how anti-MM drugs and COX inhibitors could affect PGE2 and TXB2 release and NF- κB activation, cells were treated with BRZ, DEX and THA alone or in combination with the highly selective COX-1 inhibitor mofezolac, or the selective COX-2 inhibitor celecoxib. PGE2 and TXB2 biosynthesis, and NF-κB activation in treated cells did not change respect to the basal level measured in the untreated cells (Table S1-S2).
CalceP-gp interacting profile by COX inhibitors
The possible action of the COX inhibitors on P-gp function was investigated by combining the results of three assays: 1) inhibition of the transport of a pro-fluorescent probe (Calcein-AM) in cells overexpressing P-gp, MDCK-MDR1 cells; 2) apparent permeability (Papp) determination in Caco-2 cell monolayer; 3) ATP cell depletion in MDCK-MDR1 cells.
The first assay establishes the potency of the interaction between the compounds and the efflux pump by measuring the transport inhibition of the pro-fluorescent probe Calcein-AM, which is a P-gp substrate, in a cell line overexpressing P-gp (MDCK-MDR1 cells).
The second assay measures the ratio (BA/AB) between passive diffusion (BA: flux from the basolateral compartment to the apical compartment, due to passive diffusion since P-gp is apically expressed) and the active transport (AB: flux from the apical compartment, where the pump is present, to the basolateral compartment, influenced by the P-gp-mediated active transport).
If BA/AB value is < 2, the compound can be considered an inhibitor since the compound is not actively effluxed by the apically localized pump.
A BA/AB value > 2 classifies the compound as transported P-gp substrate since the compound is actively effluxed from the pump located at the apical level.
The third assay detects the ATP consumption as a result of the transport mediated by the pump; generally, a substrate that is transported by the pump induces ATP cell depletion, whereas a P-gp inhibitor does not induce ATP consumption.
Compounds displaying a BA/AB > 2 but not inducing an ATP cell depletion belong to the class IIB3 substrates [36, 37, 41 ,42].
Since P-gp, MRP1 and BCRP are the Multi Drug Resistance related proteins involved in determining resistance to chemotherapy in cancer patients, the selectivity of COX inhibitors toward the other two MDR sister proteins, MRP1 and BCRP, was also evaluated by measuring the inhibition of the efflux of Calce in-AM (MRP1 substrate) in cells overexpressing MRP1 (MDCK- MRP1 cells) and of the fluorescent probe Hoechst 33342 (BCRP substrate) in cells overexpressing BCRP (MDCK-BCRP cells).
Mofezolac and aspirin did not interact with any efflux pump. Celecoxib and SC 560 were completely inactive towards BCRP, whereas they interacted with MRP1 (EC50 = 3.47 μM and 24.5 μM for celecoxib and SC 560, respectively). SC 560 was active towards P-gp (EC50 = 30.8 μM), while celecoxib showed a low activity towards the efflux pump (EC50 = 87 μM).
All COX inhibitors showed a category IIB3 substrate profile since Papp (BA/AB) values were > 2 for all compounds and they were all unable to induce ATP cell depletion.
The only exception was aspirin, which displayed a weak P-gp inhibitor profile having a Papp < 2, not inducing an ATP consumption and not displaying a P-gp inhibiting effect (Table 2).
COX expression and drugs effects in patients derived BMSCs
Evidences collected by the above described experiments carried out on human cell lines (hMMCLs) allowed to establish their different viability and behavior upon treatment with the same anti-MM drugs and COX inhibitors. Based on the obtained results, the anti MM drugs and COX inhibitors cytotoxicity seems to be not COX-mediated at least in the chosen hMMCLs. Furthermore, to overcome the limits of immortalized cell lines, a co-culture of primary MM plasma cell lines (CD138+) and bone marrow stromal cells (BMSCs), isolated from patients with active MM disease, was treated with the same drugs previously used to test their effect on hMMCLs. BMSCs seem to have a pro-tumor effect on MM cells [43, 44] and because it was supposed that pro-inflammatory mediators released upon COX activation could be correlated to the permissive microenvironment induced by BMSCs (Figure 4) [45] it appeared interesting to investigate COXs expression and their activity before and after the pharmacological treatment of BMSCs.
COX-1 and COX-2 were found to be expressed in the untreated bone marrow stromal cells. COX expression was almost completely suppressed upon treatment with anti-MM drugs (BRZ, DEX, THA), and the treatment of mofezolac together with DEX determined a slight COX-1 expression (Figure 5).
The cytotoxic activity on the bone marrow stromal cells of the three anti-MM drugs (BRZ, DEX and THA) alone or in combination with COX inhibitors (mofezolac, celecoxib and aspirin) was also investigated (Figure 6).
The hBMSCs treatment with anti-MM drugs determined a very low cytotoxic effect confirming that hBMSCs represent a microenvironment which favors MM cells proliferation. Therefore this condition could in part justify the low observed toxicity of anti-MM drugs on the seven hBMSCs (Figures S2-S8). This status is reversed in the presence of SC 560 and mofezolac, two highly selective COX-1 inhibitors, co-administered with THA.
Conclusions
Multiple myeloma (MM) is the second most worldwide common blood tumor, representing 10-12% of all haematological malignancies. It remains still incurable and is characterized by a great burden of morbidity, that significantly impacts on the public health system and the quality of the life of the patients. The major limitation to identify a proper MM therapy is mainly caused from a wide interindividual variation in response to the clinically available drugs. Somehow, the heterogeneous response to the treatment is mostly due to the molecular characteristics of the tumor. Based on the best of our knowledge, COX role in multiple myeloma is not well defined yet. Seven different human MM cell lines were studied to test the antiproliferative effects exerted by anti-MM drugs alone or in combination with COX inhibitors. They were selected to represent the different stage of this disease. Two of the seven cell lines that express COX-1 together with cell lines that do not express the COX-1 isoform were selected and treated with anti-MM drugs that previously shown the most representative changes in cell viability. All the collected data indicate that treatment with anti-MM drugs added with COX inhibitors has no synergistic cytotoxic effect and cells exhibited a different sensitivity to the same pharmacological treatment seeming not COX-1-mediated. These findings are congruent with the heterogeneity of the cells, originally selected as representative of the different disease conditions. This resemble what clinically happen, MM patients treated almost in the same way have a different response and prognosis.
Experimental conditions more adherent to the in vivo status were built by using stromal cells, isolated from bone marrow aspiration of patients with active MM disease. Their culture treated with the same drugs used to investigate hBMSCs behaviour revealed a drug low cytotoxic effect confirming that hBMSCs behaviour induce a microenvironment which favors MM cells viability, condition similar to the in vivo that lower antitumoral efficacy of anti-MM drugs. This status is reversed in the presence of two highly selective COX-1 inhibitors SC 560 and mofezolac when co-administered with THA.
Since the bone marrow represents a survival-promoting microenvironment of MM tumor cells, even protecting them from anticancer drugs [48-55], further studies with cell culture 3D model are ongoing to investigate whether COX inhibitors could affect the protective role of BMSCs isolated from patients with active MM.
Acknowledgments
The authors acknowledge First AIRC Grant-MFAG2015 (Project Id. 17566 to Maria Grazia Perrone) and Investigator Grant (Project Id. 20441 to Vito Racanelli) for supporting their work.
Patient Informed Consent: Protocol number: 0055223/12/07/2017/AOUCPG23/FARMPOL/P Study n° 5145.
Supplementary-Data
- Palumbo A, Anderson K (2011) Multiple myeloma. N Engl J Med 364: 1046-60.
- Murray MY, Rushworth SA, Zaitseva L, Bowles KM, Macewan DJ (2013) Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle 12: 2144-53.
- Boccadoro M, Morgan G, Cavenagh J (2005) Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int 5: 1-18.
- Martiniani R, Di Loreto V, Di Sano C, Lombardo A, Liberati AM (2012) Biological activity of lenalidomide and its underlying therapeutic effects in multiple myeloma. Adv Haematol 2012: 842945-56.
- Stewart AK, Jacobus S, Fonseca R, Weiss M, Callander NS (2015) Chanan-Khan AA et al. Melphalan, prednisone, and thalidomide vs melphalan, prednisone, and lenalidomide (ECOG E1A06) in untreated multiple myeloma. Blood 126: 1294-301.
- Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR (2008) Improved survival in multiple myeloma and the impact of novel therapies. Blood 111: 2516–20.
- Zrieki A, Farinotti R, Buyse M (2008) Cyclooxygenase inhibitors down regulate P-glycoprotein in human colorectal caco-2 cell line. Pharm. Res 25: 1991-201.
- Vitale P, Scilimati A, Perrone MG (2015) Update on SAR Studies Toward New COX-1 Selective Inhibitors. Curr. Med. Chem. 22: 4271-92.
- Perrone MG, Scilimati A, Simone L, Vitale P (2010) Selective COX-1 Inhibition: A Therapeutic Target to be Reconsidered. Curr Med Chem 17: 3769-805.
- Vitale P, Panella A, Scilimati A, Perrone MG (2016) COX-1 inhibitors: Beyond Structure Towards Therapy. Med Rev Res 36: 641-71.
- Williams TJ, Peck MJ (1977) Role of prostaglandin-mediated vasodilatation in inflammation. Nature 270: 530-2.
- Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS (2002) Prostaglandin E2, transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 8: 289-93.
- Calvello R, Panaro MA, Carbone ML, Cianciulli A, Perrone MG, et al. (2012) Novel selective COX-1 inhibitors suppress neuroinflammatory mediators in LPS-stimulated N13 microglial cells. Pharmacol Res 65: 137-48.
- Ghosh N, Chaki R, Mandal V, Mandal SC (2010) COX-2 as a Target for Cancer Chemotherapy. Pharmacol Rep 62: 233-44.
- Steinbach G, Lynch PM, Phillips RKS, Wallace MH, Hawk E, et al. (2000) The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 342: 1946-52.
- Davis TW, O’ Neal JM, Pagel MD, Zweifel BS, Mehta PP, et al. (2004) Synergy between celecoxib and radiotherapy results from inhibition of cyclooxygenase-2-derived prostaglandin E2, a survival factor for tumor and associated vasculature. Cancer Res. 64: 279-85.
- Ramon S, Woeller CF, Phipps RP (2013) The influence of Cox-2 and bioactive lipids on hematological cancers. Curr Angiogenes 2: 135-42.
- Giles FJ, Kantarjian HM, Bekele BN, Cortes JE, Faderl S, et al. (2002) Bone marrow cyclooxygenase-2 levels are elevated in chronic-phase chronic myeloid leukemia and are associated with reduced survival. Br J Haematol 119: 38-45.
- Zetterberg E, Lundberg, L. G.; Palmblad, J. Expression of Cox-2, Tie-2 and glycodelin by megakaryocytes in patients with chronic myeloid leukaemia and polycythaemia vera. Br J Haematol 2003, 121, 497-9.
- Wun T, McKnight H, Tuscano JM (2004) Increased cyclooxygenase-2 (COX-2): A potential role in the pathogenesis of lymphoma. Leuk Res 28: 179-90.
- Ladetto M, Vallet S, Trojan A, Dell’Aquila M, Monitillo L, Rosato R, et al. (2004) Cyclooxygenase-2 (COX-2) is frequently expressed in multiple myeloma and is an independent predictor of poor outcome. Blood 105: 4784-91.
- Hazar B, Ergin M, Seyrek E, Erdoǧan Ş, Tuncer I, Hakverdi S (2004) Cyclooxygenase-2 (COX-2) expression in lymphomas. Leuk Lymphoma 45: 1395-99.
- Perrone MG, Luisi O, De Grassi A, Ferorelli S, Cormio G (2004) Translational Theragnostic of Ovarian Cancer: where do we stand? Curr. Med. Chem. 27: 5675-715
- Perrone MG, Malerba P, Uddin MJ, Vitale P, Panella A, Crews BC, et al. PET radiotracer [18F]-P6 selectively targeting COX-1 as a novel biomarker in ovarian cancer: Preliminary investigation. Eur. J. Med. Chem. 2014;80:562-568.
- Scilimati A, Ferorelli S, Iaselli MC, Miciaccia M, Pati ML, et al. (2019) Targeting COX-1 by mofezolac-based fluorescent probes for ovarian cancer detection. Eur J Med Chem 179: 16-25.
- Pati ML, Vitale P, Ferorelli S, Iaselli M, Miciaccia M, et al. (2019) Translational impact of novel widely pharmacological characterized mofezolac-derived COX-1 inhibitors combined with bortezomib on human multiple myeloma cell lines viability. Eur J Med Chem164: 59-76.
- Casalino G, Coluccia M, Pati ML, Pannunzio A, Scilimati A, Perrone MG. Intelligent Microarray Data Analysis through Non-negative Matrix Factorization to Study Human Multiple Myeloma Cell lines. Appl Sci 9: 5552-70.
- Ding J, Tsuboi K, Hoshikawa H, Goto R, Mori N, et al (2006) Cyclooxygenase Isozymes Are Expressed in Human Myeloma Cells but not Involved in Anti-Proliferative Effect of Cyclooxygenase Inhibitors. Mol Carcinog 45: 250-59.
- Umezu T, Imanishi S, Yoshizawa S, Kawana C, Ohyashiki JH, (2019) Induction of multiple myeloma bone marrow stromal cell apoptosis by inhibiting extracellular vesicle miR- 10a secretion. Blood Adv 3: 3228-40.
- Samadi AK, Bilsland A, Georgakilas AG, Amedei A, Amin A, et al. (2015) A Multi- targeted Approach to Suppress Tumor-Promoting Inflammation. Semin Cancer Biol 35: S151–84.
- Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, et al. (2010) Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 116: 2543-53.
- Moreaux J, Klein B, Bataille R, Descamps G, Maïga S, et al. (2011) A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematol 96: 574-82.
- Cingolani G, Panella A, Perrone MG, Vitale P, Di Mauro G, et al. (2017) Structural basis for selective inhibition of Cyclooxygenase-1 (COX-1) by diarylisoxazoles mofezolac and 3-(5- chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6). Eur J Med Chem 138: 661-68.
- Yuan C, Smith WLA (2015) Cyclooxygenase-2-dependent Prostaglandin E2 Biosynthetic System in the Golgi Apparatus. J Biol Chem 290: 5606-20.
- International Myeloma Working Group (2003) Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br. J. Haematol. 121:749-57.
- Leone P, Di Lernia G, Solimando AG, Cicco S, Saltarella I, et al. (2018) Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. Oncoimmunology 8: e1486949-61.
- Calvello R, Lofrumento DD, Perrone MG, Cianciulli A, Salvatore, R. et al. (2017) Highly selective cyclooxygenase-1 inhibitors P6 and mofezolac counteract inflammatory state both in vitro and in vivo models of neuroinflammation. Front. Neurol. 8: 251-61.
- Riganti C, Contino M, Guglielmo S, Perrone MG, Salaroglio IC, et al. (2019) Design, Biological Evaluation and Molecular Modelling of Tetrahydroisoquinoline Derivatives: Discovery of A Potent P-glycoprotein Ligand Overcoming Multi-Drug Resistance in Cancer Stem Cells. J Med Chem 62: 974-86.
- Contino M, Guglielmo S, Perrone MG, Giampietro R, Rolando B, et al. (2018) New tetrahydroisoquinoline-based P-glycoprotein modulators: decoration of the biphenyl core gives selective ligands. Medchemcomm 9: 862-69.
- Capparelli E, Zinzi L, Cantore M, Contino M, Perrone MG, et al. (2014) SAR Studies on Tetrahydroisoquinoline Derivatives: The Role of Flexibility and Bioisosterism to Raise Potency and Selectivity toward P-Glycoprotein. J Med Chem 57: 9983-94.
- Perrone MG, Vitale P, Ferorelli S, Boccarelli A, Coluccia M, et al. (2017) Effect of mofezolac-galactose distance in conjugates targeting cyclooxygenase (COX)-1 and CNS GLUT-1 carrier. Eur J Med Chem 141: 404-16.
- Colabufo NA, Contino M, Cantore M, Capparelli E, Perrone MG, et al. (2013) Naphthlenyl derivatives for hitting P-gp/MRP1/BCRP transporters. Bioorg Med Chem 21: 1324-32.
- Castells M, Thibault B, Delord JP, Couderc B (2012) Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci 13: 9545–75.
- Liang X, Song E (2020) The role of bone marrow stromal cells in blood diseases and clinical significance as a crucial part of the hematopoietic microenvironment. Annals of Blood 5: 1-10.
- Markovina S, Callander NS (2010) Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol Cancer 9: 176-89.
- Miciaccia M, Belviso BD, Iaselli M, Cingolani G, Ferorelli S, et al. (2020) Three- dimensional structure of human cyclooxygenase (hCOX)-1. Sci Rep 2020.
- Kiefer JR, Pawiitz JL, Moreland KT, Stegeman RA, Hood WF, et al. (2000) Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 405: 97-101.
- Meads MB, Hazlehurst LA, Dalton WS (2008) The Bone Marrow Microenvironment as a Tumor Sanctuary and Contributor to Drug Resistance. Clin Cancer Res 14: 2519-26.
- Perrone MG, Santandrea E, Scilimati A, Tortorella V, Capitelli F, et al. (2004) Baker’s yeast-mediated reduction of ethyl 2-(4-chlorophenoxy)-3- oxoalkanoates intermediates for potential PPARα ligands Tetrah. Asymm. 15: 3501-10.
- Vitale P, D’Introno C, Perna F, Perrone MG, Scilimati A. (2013) Kluyveromyces marxianus CBS 6556 growing cells as a new biocatalyst in the asymmetric reduction of substituted acetophenones. Tetr. Asymm. 24: 389-94.
- Catalano A, Carocci A, Corbo F, Franchini C, Muraglia M, et al. (2008) Constrained analogues of tocainide as potent skeletal muscle sodium channel blockers towards the development of antimyotonic agents. Eur. J. Med. Chem. 43: 2535-40.
- Di Nunno L, Vitale P, Scilimati A, Simone L, Capitelli F, (2007) Stereoselective dimerization of 3-arylisoxazoles to cage-shaped bis-β-lactams syn 2,6-diaryl-3,7-diazatricyclo[4.2.0.02,5]octan-4,8-diones induced by hindered lithium amides. Tetrahedron 63:12388-395.
- Di Nunno L, Scilimati A, Vitale P, (2002) Regioselective synthesis and side-chain metallation and elaboration of 3-aryl-5-alkylisoxazoles. Tetrahedron 58: 2659-65.
- De Luca A. Talon S., De Bellis M, Desaphy J-F, Lentini G, et al. (2003) Optimal requirements for high affinity and use-dependent block of skeletal muscle sodium channel by N-benzyl analogs of tocainide-like compounds. Mol. Pharmacol. 64: 932-45.
- Di Nunno L, Scilimati A (1986) Decomposition of arylazides by THF/n-butyllithium-II-isolation of 1-aryl-4,5-dihydro-5-hydroxy-1h-1,2,3-triazoles. Tetrahedron 42:3913-39.
FIGURE 1
Figure 1: Examples of clinically used anti-multiple myeloma drugs
FIGURE 2
Figure 2: Chemical structures of some COXs inhibitors and their Selectivity Index (SI). SI corresponds to the ratio COX-2 IC50/COX-1 IC50. SI values higher than 1 means that the compound is a selective COX-1 inhibitor [8-10]
FIGURE 3
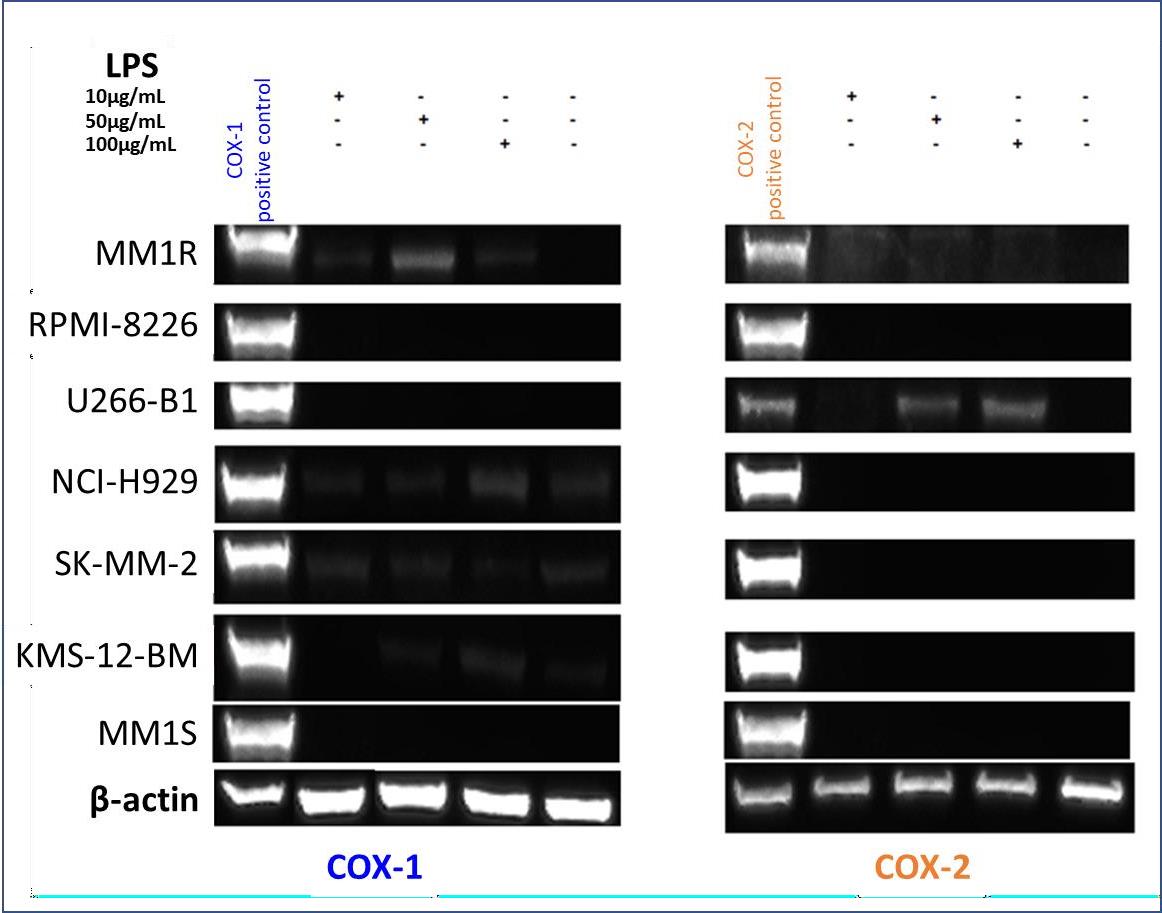
Figure 3: Adjusted image of Western blotting experiments depicting the expression of COX-1 and COX-2 isoenzymes determined by Western blotting. Original gel blots are reported in Supplementary Materials
FIGURE 4
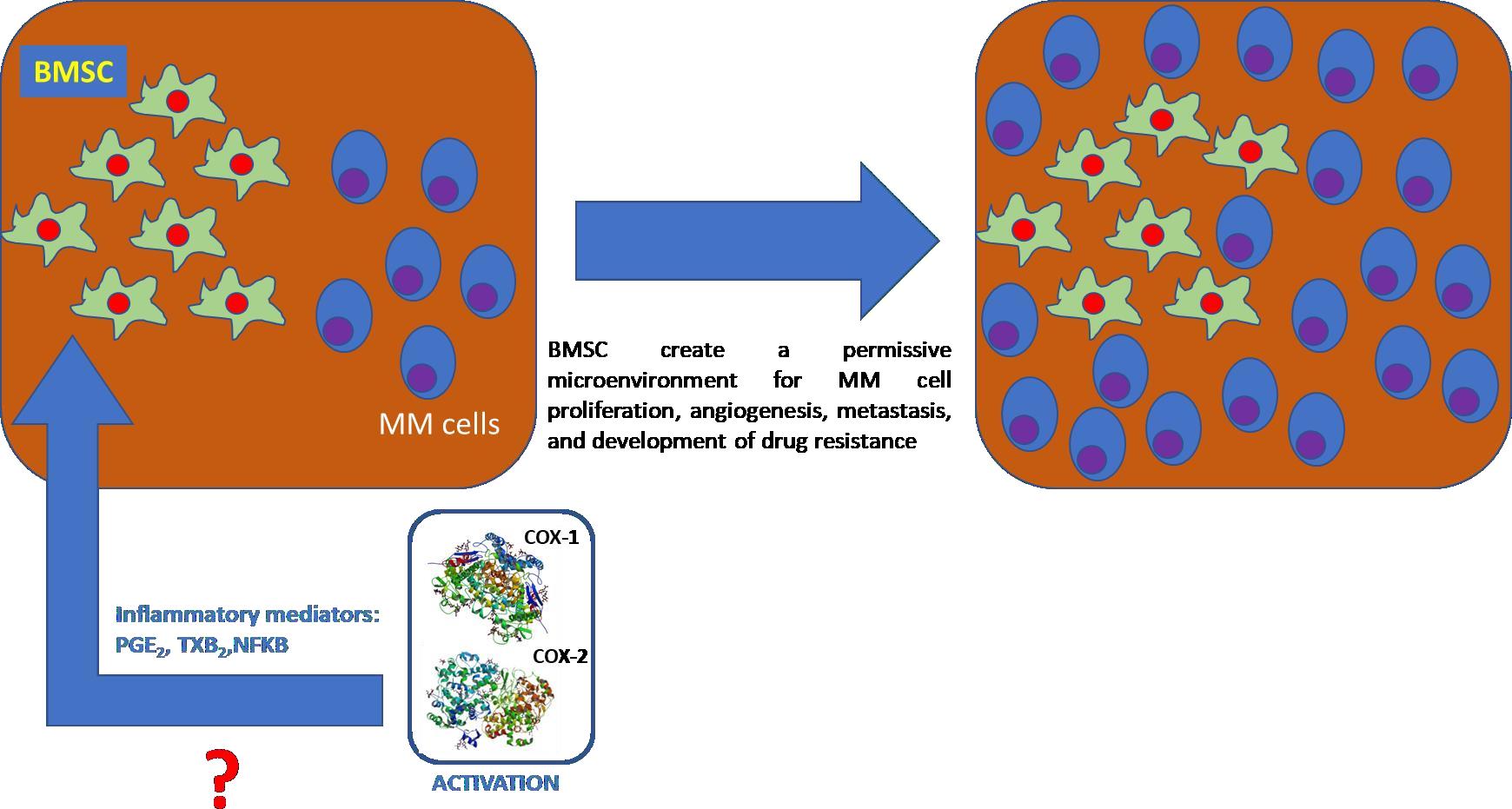
Figure 4: Hypotesis of COX activation in a BMSCs milieu, in turn permissive for MM cells proliferation, angiogenesis inducer, metastasis and drug resistrance development. COXs structures used are PDB: 6Y3C for COX-1 [46] and PDB:1CVU for COX-2 [47]
FIGURE 5

Figure 5: Expression of COXs in BMSCs upon treatment with mofezolac alone or in combination with BRZ, DEX or THA. Cells were incubated for 48 hours with (+) or without (-) 100 μM MOF, 100 μM CEL, 100 μM DEX, 100 μM THA, 300 nM BRZ. 30 μg of protein extract were applied to Western blot. The protein level of β-actin was used as the loading control. HEK-293 COX-1 cells are the positive control. Original blots are in the Supporting Materials
FIGURE 6
Figure 6: Anti-proliferative activity of anti-MM drugs alone or added with COXs inhibitors on hBMSCs growth
Tables at a glance
Figures at a glance