L-plastin Regulates Invasion and Possibly Not the Migration of Prostate Cancer (PC3) Cells
Received Date: March 30, 2020 Accepted Date: April 30, 2021 Published Date: May 03, 2021
doi: 10.17303/jcrto.2021.9.105
Citation: Sunipa Majumdar (2021) L-plastin Regulates Invasion and Possibly Not the Migration of Prostate Cancer (PC3) Cells. J Cancer Res Therap Oncol 9: 1-17.
Abstract
Metastatic cancer cells use the actin-bundling process for constant remodeling of the actin cytoskeleton for adhesion, migration, and invasion. L-plastin is an actin-bundling protein that belongs to the plastin family. L-plastin has been identified in several malignant tumors of the colon, prostate, and breast and contributes to cancer cell invasion in a phosphorylation-dependent manner. Our initial characterization in PC3 prostate cancer (PCa) cell line derived from bone metastases demonstrated L-plastin expression in PCa cells but not in other PCa cell lines tested. Hence, in this study, we aimed to identify L-plastin’s role in the migration and invasion of PC3 cells. Immunostaining analysis demonstrated a punctate distribution of L-plastin and patchy actin staining in PC3 cells with a minimal colocalization between L-plastin and actin at the invadopodia. However, L-plastin overexpression in PC3 cells increased L-plastin’s colocalization and actin at the invadopodia and during the invasion. In a wound-healing assay, these cells displayed a significant reduction in migration. L-plastin and invadopodia connections were confirmed using the L-plastin knockdown strategy in PC3 cells (PC3/Si). PC3/Si cells demonstrated an increased migration, which corresponded to punctate podosome-like structures. However, a decrease in the number of invadopodia contributed to a significant reduction in invasion. Additionally, tumorsphere formation was significantly reduced in PC3/Si cells than in PC3 cells. In conclusion, our observations suggest that L-plastin regulates the formation of invadopodia required for prostate cancer invasion. Our results highlight that it could be a novel therapeutic target for androgen-independent metastatic prostate cancer.
Keywords: L-plastin, Invadopodia; Migration; Invasion; PC3 Cells; Prostate Cancer
Introduction
Prostate cancer (PCa) is the second most commonly diagnosed cancer among men, and it is the fifth leading cause of cancer-related deaths worldwide [1]. Metastatic PCa can be highly heterogeneous, with the number of metastases being a vital predictor of cause-specific mortality Mortality due to PCa mainly results from tumor cell metastasis to distant sites, particularly the bones [3,4]. Although surgery, radiation, and androgen ablation treatments are effective against localized prostate tumors, approximately 15%–20% PCa patients develop advanced stages of the disease, leading to bone and lymph node metastases [5-7]. No effective treatment exists for advanced stages, mainly because tumor growth becomes resistant to castration [8,9].
The actin cytoskeleton's reorganization is associated with several key physiological processes, including differentiation, endocytosis, exocytosis, cell motility, and invasion [10-18]. Generally, several actin-binding and regulatory proteins modulate actin organization under physiological and pathophysiological conditions [19-25]. L-plastin (LPL) is an actin-binding (bundling) protein that has been identified as a tumor-associated protein [26-30]. It is generally expressed in the leukocytes, and it is not present in non-hematopoietic cells. It is thought to be involved in the differentiation of hematopoietic cells. However, the expression of LPL in various tumor cells suggests that it may be involved in neoplastic processes [31].
Plastins belong to a class of actin-bundling proteins. The L, T and I isoforms are tissue-specific actin-binding proteins. T-plastin is constitutively expressed in epithelial and mesenchymal cells [32], and only L- and T-plastins are involved in actin cytoskeletal reorganization [33-36]. L-plastin is expressed in 68% of epithelial carcinomas and 53% of non-epithelial mesenchymal tumors [37]. Moreover, examining various human tumor cell lines revealed that more than 90% of tumor cells display varying degrees of constitutive LPL expression [38]. LPL expression in prostatic epithelial cells is linked to the malignant state because LPL expression is not observed in normal epithelial cells or epithelium of benign prostatic hyperplasia lesions [39].
However, the functional consequences of LPL expression in PCa are unknown. Therefore, in this study, we aimed to identify the expression levels of LPL in different cell lines and choose a cell line expressing LPL to elucidate its possible functional significance in prostate cancer progression. Here, we provide compelling evidence that LPL expression is higher in bone metastatic PC3 cells than in other cancer cell lines and benign hyperplasic and normal prostatic epithelial cells. LPL overexpression in PC3 PCa cells increased invasion, whereas LPL knockdown in PC3 cells reduced the invasion capability.
Material and Methods
Reagents
Antibodies against LPL (SC-16657; D-16) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against GAPDH (anti-rabbit, catalog no. G9545), mitomycin C, the cell proliferation assay (MTT) kit, and other chemicals, in general, were purchased from Sigma-Aldrich (MO, USA). Matched normal tissue and tumor tissue lysates from a single person were purchased from Abcam (Cambridge, MA, USA). LPL-small interfering RNA (siRNA) and control siRNA were purchased from GE Healthcare (Uppsala Sweden) and Ambion Thermo Fisher Scientific (MA, USA), respectively. The FITC-conjugated secondary antibody was purchased from Jackson Immunoresearch (PA, USA).
Cell culture and cell lines
Prostate cancer cells, such as PC3, LNCaP, DU145, and benign prostatic hyperplasic (BPH) cells, were maintained in RPMI 1640 (Gibco BRL, Life Technologies, Bethesda, MD, USA) containing 10% fetal bovine serum (FBS; Gibco, NY, USA) and 1% penicillin/streptomycin [40]. PC3 cells were derived from skeletal metastases, LNCaP from lymph node metastasis, and DU-145 from brain metastasis. All cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). BPH cells were used as controls. PC3 cells stably expressing LPL or with a transient knockdown of LPL were produced as described below.
Generation of stable L-plastin-overexpressing PC3 cells
The vector plasmid pCEP-4 (Thermo Fisher Scientific (MA, USA) with an LPL cDNA insert and without any insert was transfected into PC3 cells using Lipofectamine 2000 (Invitrogen, NY, USA), according to the instructions of the manufacturer. Following transfection, cells were maintained in G418 sulfate (Sigma-Aldrich, MO, USA), and individual clones (30 clones) were isolated as previously described [40]. LPL expression levels were measured in 26 individual clones, and the results are provided in the supplementary Figure. The clone (clone 26), which expressed the highest levels of LPL, was amplified and used for further studies. PC3 cells with overexpression of LPL are denoted as PC3/LPL.
Knockdown of L-plastin in PC3 cells using a siRNA
siRNA against LPL (Smart pool) and control (denoted as scrambled) were purchased from Dharmacon and Ambion, respectively. According to the manufacturer's protocol, 100 nm siRNA was transfected using Lipofectamine 2000, as previously described [3,41]. After 36–48 h, the lysates were prepared and subjected to immunoblotting with an antibody against LPL. The PC3 cells with LPL knockdown are denoted as PC3/Si. PC3 cells transfected with a control scrambled siRNA were used as controls, and they are represented as PC3/Sc.
Immunoblotting analyses
Equal amounts of lysate proteins were used for immunoblotting, as previously described [42]. Briefly, equal amounts of proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride filter membranes (PVDF; Sigma MO, USA) for immunoblotting. Membranes were blocked with 5% milk in phosphate-buffered saline containing 0.5% Tween (PBST) for 2 h, and they were incubated with a primary antibody of interest (e.g., LPL at 1:1000 dilution) at 4 °C overnight. After washing thrice with PBST for 10 min each, the membranes were incubated with a species-specific secondary antibody conjugated to horseradish peroxidase (1:5000 dilution) at room temperature (RT; around 20-220C) for 1 h. After washing thrice with PBST for 10 min each, the protein bands were visualized using chemiluminescence using an ECL kit (Millipore Sigma MO, USA).
Immunostaining and actin staining of cells
Immunostaining and actin staining were performed as previously described [15,43]. PC3 and PC3/LPL cells were cultured on coverslips in a 6-well dish at 37 °C for 14–16 h. Cells were washed three times with PBS at RT, and they were fixed in 4% formaldehyde in PBS for 10 min. After washing thrice with PBS, cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min and blocked with 5% donkey serum in PBST for 1 h. Between steps, the cells were washed with PBST. After washing thrice with PBST, cells were immunostained with an LPL antibody (1:100 dilution) in 5% goat serum at 4 °C overnight, followed by incubation with FITC-conjugated (1:1000 dilution) species-specific secondary antibody for 3-4h. Subsequently, cells stained for LPL were stained with rhodamine-phalloidin (1:500 dilution in PBS) for actin staining, as previously described [15]. The coverslips were washed and mounted on glass slides containing a mounting solution (Vector Laboratories, CA, USA ); subsequently, coverslips were sealed with clear nail polish around the coverslips' edge. The immunostained cells were viewed and photographed using Zeiss LSM 510 Meta confocal microscope (Hercules, CA, USA). Images were saved in TIF image format, and they were processed using the Adobe Photoshop software program (Adobe Systems, Inc., Mountain View, CA, USA).
Immunohistochemistry (IHC) assay
Tissue microarray (TMA) sections containing adenocarcinoma (grades 1–4) and prostate cancer cells adjacent to the normal cells were purchased from US Biomax, Inc. (Rockville, MD, USA). The sections were arranged in duplicate for each case. Goat polyclonal LPL (D-16) antibody (Santacruz, CA, USA) was used for IHC analysis, and the TMA sections were processed, stained, and analyzed as previously described [3]. Images were taken with an Aperio ScanScope CS system (Vista, CA, USA).
Wound-healing assay
PC3, PC3/LPL, and PC3/Si cells were cultured in a 6-well plate to near confluency, as previously described [44]. Scratches were made with a 10 μL pipette tip (~25–30 μm in width), and the cells were immediately washed with RPMI medium containing 10% FBS to remove the detached cells. Mitomycin (10 μg/mL) was added to the medium to inhibit cell proliferation. Hence, the observed increase in wound healing was due to an augmentation of migration and not cell multiplication. Cell migration was monitored for 18 h, and pictures were taken at 0 h and 18 h using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay The MTT assay was performed as previously described [45] and as per the manufacturer's instructions. Briefly, PC3, PC3/LPL, and PC3/Si cells were seeded at a cell density of 2000–3000 cells/well in a 96-well plate for 16 h. MTT was added to each well, and the plate was incubated at 37 °C for 2 h. The MTT solubilization solution was added to the wells to stop the chemical reaction, and the absorption was measured at 570 nm. The percent proliferation was expressed in different cell lines with the subtraction of background absorbance.
Fluorescent gelatin degradation assay to determine the invasion capability of cells
Cross-linked-fluorophore (FITC)-conjugated gelatin matrices were coated on coverslips as previously described [46]. The coverslips were rinsed thrice with PBS, and they were stored at 4 °C until use. Before plating the cells, the gelatin substrates were quenched by incubating with RPMI containing 10% FBS (500 μL/well) at 37 °C for 30–60 min. PC3, PC3/LPL, and PC3/Si cells were seeded at a cell density of 2000–3000 cells/well to evaluate invadopodia formation and invasion capability. The cells were incubated for 16 h and washed with PBS. The cells were then fixed with 3.7% formaldehyde (250–500 μL/well) at RT for 30 min, and they were stained with rhodamine-phalloidin (1:500 dilution in PBS; Sigma) for 2 h or at 4 °C overnight. Cells were washed thrice with PBS, mounted on a glass slide with a mounting solution (Vector Laboratories, Inc., Burlingame, CA, USA), and sealed with a nail polish [43,47]. Gelatin matrix and actin-stained cells were viewed and photographed using a Nikon Spinning confocal microscope with 510 LSM Meta Software. Images were saved in the TIF image format, and they were processed using Adobe Photoshop.
Tumorsphere formation assay
The tumorsphere formation assay was performed according to the manufacturer's protocol (R&D system, Minneapolis, MS) [48,49]. Briefly, cells were plated in limited numbers in a six-well ultralow adhesion culture plate, and they were cultured for 7 days to induce tumorsphere formation. After 7 days of culture, tumorsphere images were taken using an image reader (Cytation 3, Biotek, Winooski, VT). Images were processed using Adobe Photoshop.
Results
LPL is highly expressed in human prostate cancer, tumor tissue lysates, and human prostatic adenocarcinoma tissues
Using cell lines and tumor tissue lysates from PCa cells, we sought to determine the expression of LPL. We used PCa cells derived from multiple metastases as indicated below: PC3 cells from the bone, DU145 cells from the brain's dura mater, and LNCaP cells from lymph node metastases. BPH cells were used as controls. Immunoblotting with an LPL antibody demonstrated an increased LPL expression in PC3 cells (Figure 1A, lane 2) than in the other tested cell lines (lanes 1, 3, and 4). The expression levels in the indicated cell lines from three different experiments are shown in the graph (bottom panel in Figure 1A). PC3 cells expressed more LPL proteins, as demonstrated by others [29,30].
Next, we performed the immunoblotting analysis of total lysates prepared from normal prostatic tissues (NT) and tumor tissues (TT) with an antibody against LPL (Figure 1B). LPL expression was higher in the lysates prepared from TT (Figure 1B, lane 2) than from NT (Figure 1B, lane 1). To further validate the immunoblotting findings, we performed IHC analysis with an antibody against LPL in a human prostate cancer TMA purchased from Biomax (Figure 1; panels C and D). A lower magnification view of a representative microarray panel containing NT and TT sections (24 cores) is shown in Figure S1. Arrays stained for LPL and non-immune serum are shown in Figure S1. Arrows in Figure S1 point to the sections shown in the medium (C and D) and higher (C' and D') magnification views in Figure 1. The LPL stained sections presented in Figure 1 (C and C') demonstrate normal luminal epithelial cells, stromal cells, and adenocarcinoma (indicated by an arrow in C) adjacent to the lumen (Figure 1C'). LPL staining was very low in luminal epithelial and basal cells (indicated by asterisks in C') compared to the intense LPL staining in adenocarcinoma, which was adjacent to the lumen.
Similarly, an intense LPL staining was observed in the adenocarcinoma sections at stages and IV. No distinct lumen was observed in this section, and a random distribution of carcinoma cells was observed (Figures 1D and D'). The relative distribution of LPL in the immunostained TMA sections was semi-quantitatively analyzed by two other investigators, and the results are presented in Table 1. LPL expression was higher in prostatic adenocarcinoma than in luminal epithelial cells, similar to previous results [39].
A stable LPL overexpression reduces cell migration while LPL knockdown enhances cell migration
To determine the functional importance of LPL in PC3 cells, we used an LPL overexpression and knockdown strategy. Using LPL full-length plasmids previously generated in our lab [50, 51], we overexpressed the full-length LPL plasmid in PC3 cells. A significant increase in the expression level of LPL was observed in PC3 cells transfected with full-length LPL (PC3/LPL) constructs (Figure 2A, lane 2, and Figure S2) than in PC3 cells alone (Figure 2A, lane1). We generated approximately 26 individual clones to determine LPL expression. The expression levels of standard LPL in clonal isolates are shown in Figure S2. Among the individual clones generated and tested for LPL expression level, we used one clone (clone #26), demonstrating the maximum overexpression of LPL (Figure S2, lane 27) for further studies.
Next, to determine the functional consequence of LPL overexpression (Figure 2A) on cell migration in PC3 cells, we performed a wound-healing assay (Figure 2B). Wound closure was monitored for 18 hours (Figure 2B). PC3/LPL cells displayed a significant decrease in cell migration (Figure 2B, right panel) and wound closure capabilities compared to PC3 cells (Figure 2B, left panel). Cells were pretreated with mitomycin C to ensure that the observed cell migration results were independent of cell proliferation. The wound-healing assay showed that overexpression of LPL reduced the migration ability of PC3 cells.
To further confirm whether the expression of LPL is associated with cell migration, we reduced LPL expression using siRNA against LPL in PC3 cells. SMARTpool siRNA was used to minimize the expression of LPL in PC3 cells. Immunoblotting analyses demonstrated that LPL siRNA (100nM) significantly reduced the expression of LPL in PC3 cells (PC3/Si; Figure 2C, lane as compared to untransfected (lane 1) and scrambled RNAi-transfected (PC3/Sc; lane 2) control cells. The relative expression levels from three different blots for PC3 cells, PC3/Sc, and PC3/Si cells, are shown in a graph (Figure 2D).
Interestingly, LPL knockdown increased cell migration in the wound healing assay (Figure 2E). Wound healing was monitored for 18 h. A significant reduction in cell migration was observed in PC3/Si cells (Figure 2E, right panel) than in scrambled RNAi (Sc)-transfected cells (Figure 2E, left panel). Phase-contrast microscopy of PC3, PC3/LPL, and PC3/Si is shown in Figure S3. PC3 cells overexpressing LPL (PC3/LPL) appear more rounded and less adhesive than PC3 cells. PC3/Si cells looked marginally larger than PC3/LPL or PC3 cells (Figure S3).
Overexpression of LPL increase the number of invadopodia and invasion processes in PC3 cells
We sought to determine the actin distribution in cells using rhodamine-phalloidin staining (red) and confocal microscopy analysis (Figure 3). The LPL distribution was also studied using immunostaining analysis with an LPL antibody (green). Invadopodia were counted in ~150-200 cells from three different experiments, and the results are provided in a graph (Figure 3D). PC3 and PC3/LPL cells displayed areas of actin (red) enrichment in the plasma membrane. Consistent with our previous studies [15], PC3 cells demonstrated actin-rich invadopodia-like structures (indicated by arrowheads in A and B). These structures were denser and more abundant in PC3/LPL cells (Figure 3, panels B and D). F-actin enrichment was observed in the central cytoplasmic region and plasma membrane of PC3/LPL cells compared to parental PC3 cells (Figure 3, actin). Consistent with the results of immunoblotting analysis (Figures 2C and D), LPL staining was high in PC3/LPL cells (Figure 3B; L-plastin and overlay panels), and PC3/Si cells displayed a dramatic decrease in LPL staining (Figure 3C; L-plastin and overlay panels). Colocalization (yellow) of LPL with actin was observed in areas where invadopodia were found in PC3/LPL cells (Figure 3B; overlay). Interestingly, PC3/Si cells exhibited punctate podosome-like structures (Figure 3C actin; indicated by arrows), which may explain the increased migration in wound healing assay (Figure 2E, Si panel). Correspondingly, a decrease in the number of invadopodia was observed in these cells (Figure 3D). Control vector-transfected cells (PC3/V) for LPL overexpression and scrambled RNAi-transfected PC3/Si cells are shown in Figure S4. The number of invadopodia was observed in the following order: PC3/LPL>PC3>>PC3/siRNA (Figure 3D). These observations suggest that LPL plays a role in actin-bundling or organization processes involved in invadopodia formation. Our future studies will identify whether LPL has direct or indirect role in invadopodia formation. If so, what is the mechanism by which LPL regulates invadopodia formation, and how podosomal organization occurs in the absence of LPL?
Overexpression of LPL increases the invasion of PC3 cellsInvasion is a crucial process in cancer metastasis, and invadopodia play a vital role in cancer cell invasion [11,15,16,52,53]. Previous studies have shown that invadopodia form a focal degradation of the gelatin matrix in vitro [15,16]. This study also used the in vitro gelatin degradation invasion assay to determine the invasive properties of PC3, PC3/LPL, and PC3/ Si cells, as previously described [15,46]. Cells were plated onto coverslips pre-coated with FITC-conjugated gelatin matrix (green), and they were incubated at 37 °C for 16-18 h. Confocal microscopy analysis was performed after staining the cells with rhodamine-phalloidin for actin distribution (red, Figure 4). Gelatin degradation was found underneath PC3 and PC3/LPL cells. Black holes represented the degraded matrix. An increase in the number of invadopodia in PC3/LPL cells (Figure 3D) indicated their invasive nature (Figure 4B; green). The depth and surface area of degradation were greater in PC3/LPL cells (Figure 4B; gelatin) than in PC3 and PC3/Si cells. A decrease in the number of invadopodia in PC3/LPL(Si) cells (Figure 3D) corresponds to the substantially reduced level of degradation of the gelatin matrix (Figure 4C; gelatin), which suggests that LPL plays a role in the formation of invadopodia and the polarized invasion of cells into matrix boundaries. Podosome-like structures present in PC3/Si cells (Figure 3C, actin) have no cell invasion role. The number of invadopodia (Figure 3D) and gelatin degradation (Figure 4) were in the following order: PC3/LPL>PC3>>PC3/Si.
Knockdown of LPL reduces tumorsphere formation of PC3 cells
Cancer stem cells (CSCs) can form tumorspheres in vitro when plated in low numbers with serum-free media and growth factors. CSCs result in treatment failure, thereby leading to tumor recurrence [48,54]. To determine the role of LPL in tumorsphere formation in vitro, we used PC3 and PC3/Si cells. PC3 control cells displayed more tumorspheres (Figure 5A) than PC3/Si cells (Figure 5B). A decrease in tumorsphere formation in PC3/Si cells was not due to reduced PC3/Si cell proliferation. The proliferation assay (MTT assay) demonstrated that the proliferation of PC3/LPL and PC3/Si cells was equal to that of PC3 cells (Figure S5). LPL may play an essential role in cancer cell self-renewal and differentiation, which is a part of the transformative process of metastasis and epithelial-mesenchymal transition, leading to tumor recurrence and treatment failure.
Discussion
L-plastin has a role in innate and adaptive immune cells [55,56]. LPL is expressed physiologically in hematopoietic cells, and it has been shown to regulate T-cell activation and function [19,36,56-58]. When cells are transformed malignantly, LPL is expressed abundantly non-physiologically and pathologically, under which conditions it could serve as a tumor marker [28,59-61]. Sixty-eight percent of the cancers arising from epithelial tissues express LPL [62]. The transcription factor AP4, regulated by the PI3-K/AKT pathway signaling pathway, promotes prostate cancer metastasis by upregulating the in vitro and in vivo LPL expression [63].
Previous studies have reported the role of LPL in cancer invasion and migration in PCa [29,30,63,64]. Studies have shown that the thiol oxidation of LPL on Cys101 forms a disulfide bridge with Cys42. In LPL, this process reduces the actin-based functions of tumor cells in migration, invasion, and extracellular matrix degradation. We have previously shown that LPL phosphorylation on Ser5 and 7 aa residues regulate the role of LPL in osteoclast sealing ring formation and bone resorption [35,50]. LPL expressed in PC3 cells was also phosphorylated on serine residues (data not shown). Similarly, other studies have shown that ectopic expression and phosphorylation of LPL pronounced the in vitro migration/invasion potential and in vivo metastatic capacity of human melanoma and PCa cells [28,29]. However, the role of LPL in invasion and invadopodia formation requires further investigation.
In this study, we showed that LPL expression is minimal in LNCaP and DU145 cells. LPL expression is relatively high in PC3 cells, in addition to its invasive properties due to the presence of invadopodia [15]. Therefore, we used PC3 cells in our study. Here, we used LPL overexpression and knockdown strategies in PC3 cells to analyze their roles in invasion and migratory processes systematically. As described previously [15], we used the gelatin degradation assay together with actin staining of cells to study the invasive abilities of PC3, PC3/LPL, and PC3/Si cells. LPL overexpression increased cell invasion as a result of the formation of invadopodia. Invasive migration and proteolytic degradation of the extracellular matrix (ECM) and remodeling of ECM are interdependent processes that regulate the invasion and migration of cancer cells. Previous studies have used invasion assay chambers to study the invasive property of PC3 cells with LPL knockdown [64]. Our results align with those of previous studies, which suggest that the invasion of PC3/Si cells is considerably reduced compared to that of control PC3 cells [64]. The decrease was not related to cell proliferation because the proliferation of PC3/LPL and PC3/Si cells was greater than that of PC3 control cells. Additionally, the proliferation of PC3/Si and PC3/LPL cells was comparable. In comparison, the migration was shown to be decreased in LPL knockdown PC3 cells by the same group [64], but we observed an increase in PC3/Si migration cells in the wound healing assay. The formation of podosome-like structures may have a role in migration in PC3/Si cells.
In vitro wound healing assays do not require matrix degradation. In the normal woundhealing process in vivo, protease breakdown damages extracellular matrix proteins to form new tissues. The migration and healing processes depend on the matrix molecules and chemotactic factors released by the cells subjected to migration into the scratched wound and the damaged cells during scratching of the wound [65]. Interestingly, an increase in migration was not observed in PC3/Si cells plated on the gelatin matrix, even with the formation of punctate podosome-like structures. Our previous studies on osteoclasts showed that the presence of podosomes at the leading edge increases the migration and degradation of the gelatin matrix. The track of a cleared area of gelatin matrix was due to the presence of MMP-9 in the podosomes [15]. MT1-MMP localized on podosome protrusions has been reported to mediate ECM degradation [66-68].
Podosomes coordinate cell adhesion, substratum-rigidity sensing, and matrix degradation [69-71]. Invasion requires both migration and degradation of ECM, in addition to the formation of invadopodia. We have demonstrated the possible role of Src kinase, cortactin, and WASP in the formation of invadopodia and invasion in PC3 cells. In future studies, we aim to identify the precise signaling mechanism by which the LPL mediated actin-bundling process is regulated to provide a novel therapeutic target to curtail prostate cancer progression. Failure to form a degraded matrix suggests that the podosome-like structures present in PC3/Si cells may not have any proteolytic activity. Further studies will be necessary to elucidate the migration mode of PC3/Si cells in a wound-healing assay and clarify whether these cells possess the characteristic mesenchymal motility. This study's limitation is the lack of elucidation of the molecular mechanism underlying the regulation of invadopodia formation in androgen-independent PC3 cells. Our future studies will focus on the possible molecular mechanisms associated with invadopodia formation in PC3 and PC3/LPL cells and podosome formation in the absence of LPL in PC3/Si cells. Understanding the molecular mechanism may also provide a novel molecular target for developing an effective therapeutic strategy to target key molecules involved in invadopodia formation and invasive processes.
Conclusion
Tumor cell migration and invasion are complex, and they require highly coordinated actin remodeling processes, which involve actin-binding/bundling and regulatory proteins. Actin bundling is a crucial process in invadopodia formation, invasion, and ECM degradation via the localization of matrix metalloproteases in the invadopodia region. Based on previous and present results, LPL can be an essential tumor biomarker. Cancer screening and diagnostic methods for evaluating the expression of plastins as biomarkers have been developed (72). A decrease in the invasion of PC3 cells transfected with a siRNA against LPL suggests that an antisense delivery approach can be used to obstruct the invasion and progression of PCa. We have recently shown the critical role of LPL in sealing ring formation and bone resorption in osteoclasts [35,50,73-75]. The present observations suggest that the role of LPL-mediated actin modulation is not limited only to sealing ring formation and bone resorption in osteoclasts but also to invadopodia formation. Therefore, LPL may be an essential component of invadopodia required for the invasion process and a potential therapeutic target for metastatic prostate cancer.
Acknowledgments
We appreciate the help provided by Dr. Joseph Mauban at Confocal Microscopy Core at the University of Maryland, Baltimore School of Medicine in using the Nikon scanning disk microscope; Editage (www.editage.com) for English language editing. This research was funded by the National Institute of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases (5R01AR066044) to MAC. The funders had no role in the study design, collection, analyses, or interpretation of data, writing the manuscript, or publishing the results.
Supplemental Figures
- Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7-30.
- Carter HB, Coffey DS (1990) The prostate: an increasing medical problem. Prostate 16: 39-48.
- Gupta A, Cao W, MA C (2012) Integrin avb3 and CD44 pathways in metastatic prostate cancer cells support osteoclastogensis via RUNX2/Smad5/RANKL signaling axis 2012: 1-18.
- Senbanjo LT, Chellaiah MA (2017) CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol 5: 18.
- Pratap J, Lian JB, Stein GS (2011) Metastatic bone disease: role of transcription factors and future targets. Bone 48: 30-6.
- Lattouf JB, Fadlallah H, Saad F (2011) Androgen deprivation and bone. Curr Osteoporos Rep 9: 20-4.
- Senbanjo LT, Aljohani H, Majumdar S, Chellaiah MA (2019) Characterization of CD44 intracellular domain interaction with RUNX2 in PC3 human prostate cancer cells. Cell Commun Signal 17: 80.
- McKay RR, Silver R, Bhak RH, Korves C, Cheng M, et al. (2020) Treatment of metastatic castration resistant prostate cancer with radium-223: a retrospective study at a US tertiary oncology center. Prostate Cancer Prostatic Dis 2020.
- Jeter CR, Liu B, Lu Y, Chao HP, Zhang D, et al. (2016) NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov 2: 16041.
- Jeter CR, Yang T, Wang J, Chao HP, Tang DG (2015) Concise Review: NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions. Stem Cells 33: 2381-90.
- Yamaguchi H, Pixley F, Condeelis J (2006) Invadopodia and podosomes in tumor invasion. Eur J Cell Biol. 85: 213-8.
- Teitelbaum SL. The osteoclast and its unique cytoskeleton. Ann N Y Acad Sci 1240: 14-7.
- Bretscher A, Reczek D, Berryman M (1997) Ezrin: a protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J Cell Sci 110: 3011-8.
- Nishimura Y, Yoshioka K, Bernard O, Himeno M, Itoh K (2004) LIM kinase 1: evidence for a role in the regulation of intracellular vesicle trafficking of lysosomes and endosomes in human breast cancer cells. Eur J Cell Biol 83: 369-80.
- Desai B, Ma T, Chellaiah MA (2008) Invadopodia and matrix degradation: a new property of prostate cancer cells during migration and invasion. J Biol Chem 283: 13856-66.
- Chellaiah MA (2014) CD44-Src signaling promotes invadopodia formation in prostate cancer (PC3) cells. OA Cancer (Mol Oncology) 2014: 1-6.
- Vignjevic D, Montagnac G (2008) Reorganisation of the dendritic actin network during cancer cell migration and invasion. Semin Cancer Biol 18: 12-22.
- Yamaguchi H, Condeelis J (2007) Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773: 642-52.
- Delanote V, Vandekerckhove J, Gettemans J (2005) Plastins: versatile modulators of actin organization in (patho)physiological cellular processes. Acta Pharmacol Sin 26: 769-79.
- Ujfalusi Z, Kovacs M, Nagy NT, Barko S, Hild G, et al. (2012) Myosin and tropomyosin stabilize the conformation of formin-nucleated actin filaments. J Biol Chem 287: 31894-904.
- Popowicz GM, Schleicher M, Noegel AA, Holak TA (2006) Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci 31: 411-9.
- Winder SJ, Ayscough KR (2005) Actin-binding proteins. J Cell Sci 118: 651-4.
- Giganti A, Plastino J, Janji B, Van TM, Lentz D, et al. (2005) Actin-filament cross-linking protein T-plastin increases Arp2/3-mediated actin-based movement. J Cell Sci 118: 1255-65.
- Kell MJ, Riccio RE, Baumgartner EA, Compton ZJ, Pecorin PJ, et al. (2018) Targeted deletion of the zebrafish actin-bundling protein L-plastin (lcp1). PLoS ONE 13: e0190353.
- Weed SA, Parsons JT (2001) Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene 20: 6418-34.
- Leavitt J, Chen ZP, Lockwood CJ, Schatz F (1994) Regulation of synthesis of the transformation-induced protein, leukocyte plastin, by ovarian steroid hormones. Cancer Res 54: 3447-54.
- Foran E, McWilliam P, Kelleher D, Croke DT, Long A (2006) The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int J Cancer 118: 2098-104.
- Klemke M, Rafael MT, Wabnitz GH, Weschenfelder T, Konstandin MH et al. (2007) Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. Int J Cancer 120: 2590-9.
- Riplinger SM, Wabnitz GH, Kirchgessner H, Jahraus B, Lasitschka F, et al. (2014) Metastasis of prostate cancer and melanoma cells in a preclinical in vivo mouse model is enhanced by L-plastin expression and phosphorylation. Mol Cancer 13: 10.
- Lin CS, Lau A, Yeh CC, Chang CH, Lue TF (2000) Upregulation of L-plastin gene by testosterone in breast and prostate cancer cells: identification of three cooperative androgen receptor-binding sequences. DNA Cell Biol 19: 1-7.
- Lin CS, Aebersold RH, Kent SB, Varma M, Leavitt J (1988) Molecular cloning and characterization of plastin, a human leukocyte protein expressed in transformed human fibroblasts. Mol Cell Biol 8: 4659-68.
- Arpin M, Friederich E, Algrain M, Vernel F, Louvard D (1994) Functional differences between L- and T-plastin isoforms. J Cell Biol 127: 1995-2008.
- Lin CS, Lau A, Lue TF (1998) Analysis and mapping of plastin phosphorylation. DNA Cell Biol 17: 1041-6.
- Zhou JY, Szasz TP, Stewart-Hutchinson PJ, Sivapalan J, Todd EM, et al (2016) L-Plastin promotes podosome longevity and supports macrophage motility. Mol Immunol 78: 79-88.
- Ma T, Sadashivaiah K, Chellaiah MA (2010) Regulation of sealing ring formation by L-plastin and cortactin in osteoclasts. J Biol Chem 285: 29911-24.
- Morley SC (2013) The actin-bundling protein L-plastin supports T-cell motility and activation. Immunol Rev. 256: 48-62.
- Lin CS, Chen ZP, Park T, Ghosh K, Leavitt J (1993) Characterization of the human L-plastin gene promoter in normal and neoplastic cells. J Biol Chem 268: 2793-801.
- Park T, Chen ZP, Leavitt J (1994) Activation of the leukocyte plastin gene occurs in most human cancer cells. Cancer Res 54: 1775-81.
- Zheng J, Rudra-Ganguly N, Miller GJ, Moffatt KA, Cote RJ, et al. (1997) Steroid hormone induction and expression patterns of L-plastin in normal and carcinomatous prostate tissues. Am J Pathol 150: 2009-18.
- Desai B, Rogers MJ, Chellaiah MA (2007) Mechanisms of osteopontin and CD44 as metastatic principles in prostate cancer cells. Mol Cance. 6: 18.
- Chellaiah MA, Kuppuswamy D, Lasky L, Linder S (2007) Phosphorylation of a Wiscott-Aldrich syndrome protein-associated signal complex is critical in osteoclast bone resorption. J Biol Chem 282: 10104-16.
- Chellaiah M, Fitzgerald C, Filardo EJ, Cheresh DA, Hruska KA (1996) Osteopontin activation of c-src in human melanoma cells requires the cytoplasmic domain of the integrin av subunit. Endocrinology 137: 2432-40.
- Chellaiah M, Soga N, Swanson S, McAllister S, Alvarez U, et al. (2000) Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J Biolog Chem 275: 11993-2002.
- Samanna V, Wei H, Ego-Osuala D, Chellaiah MA (2006) Alpha-V-dependent outside-in signaling is required for the regulation of CD44 surface expression, MMP-2 secretion, and cell migration by osteopontin in human melanoma cells. Exp Cell Res 312: 2214-30.
- Aljohani H, Senbanjo LT, Chellaiah MA (2019) Methylsulfonylmethane increases osteogenesis and regulates the mineralization of the matrix by transglutaminase 2 in SHED cells. PLoS ONE 14: e0225598.
- Bowden ET, Coopman PJ, Mueller SC (2001) Invadopodia: unique methods for measurement of extracellular matrix degradation in vitro. Methods Cell Biol 63: 613-27.
- Chellaiah M, Fitzgerald C, Alvarez U, Hruska K (1998) C-src is required for stimulation of gelsolin associated PI3-K. J Biolog Chem 273: 11908-16.
- Srinivasan D, Senbanjo L, Majumdar S, Franklin RB, Chellaiah MA (2018) Androgen receptor expression reduces stemness characteristics of prostate cancer cells (PC3) by repression of CD44 and SOX2. J Cell Biochem.
- Shaheen S, Ahmed M, Lorenzi F, Nateri AS (2016) Spheroid-Formation (Colonosphere) Assay for in Vitro Assessment and Expansion of Stem Cells in Colon Cancer. Stem Cell Rev 12: 492-9.
- Chellaiah MA, Ma T, Majumdar S (2018) L-plastin phosphorylation regulates the early phase of sealing ring formation by actin bundling process in mouse osteoclasts. Exp Cell Res 372: 73-82.
- Chellaiah MA, Ma T, Majumdar S (2018) L-plastin phosphorylation regulates the early phase of sealing ring formation by actin-bundling process in mouse osteoclasts. Exp Cell Res 372: 73-82.
- Weaver AM (2006) Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis 23: 97-105.
- Buccione R, Orth JD, McNiven MA (2004) Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol 5: 647-57.
- Klarmann GJ, Hurt EM, Mathews LA, Zhang X, Duhagon MA, et al. (2009) Invasive prostate cancer cells are tumor-initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis 26: 433-46.
- Wang C, Morley SC, Donermeyer D, Peng I, Lee WP, et al. (2010) Actin-bundling protein L-plastin regulates T cell activation. J Immunol 185: 7487-97.
- Wabnitz G, Balta E, Samstag Y (2017) L-plastin regulates the stability of the immune synapse of naive and effector T-cells. Adv Biol Regul 63: 107-14.
- Morley SC (2012) The actin-bundling protein L-plastin: a critical regulator of immune cell function. Int J Cell Biol 2012: 935173.
- Ishida H, Jensen KV, Woodman AG, Hyndman ME, Vogel HJ (2017) The Calcium-Dependent Switch Helix of L-Plastin Regulates Actin Bundling. Sci Rep 7: 40662.
- Goldstein D, Djeu J, Latter G, Burbeck S, Leavitt J (1985) Abundant synthesis of the transformation-induced protein of neoplastic human fibroblasts, plastin, in normal lymphocytes Cancer Res 45: 5643-7.
- Lapillonne A, Coue O, Friederich E, Nicolas A, Del Maestro L, et al. (2000) Expression patterns of L-plastin isoform in normal and carcinomatous breast tissues. Anticancer Res 20: 3177-82.
- Li J, Zhao R (2011) Expression and clinical significance of L-plastin in colorectal carcinoma. J Gastrointest Surg. 15: 1982-8.
- Lin CS, Park T, Chen ZP, Leavitt J (1993) Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J Biol Chem 268: 2781-92.
- Chen C, Cai Q, He W, Lam TB, Lin J, Zhao Y, et al. (2017) AP4 modulated by the PI3K/AKT pathway promotes prostate cancer proliferation and metastasis of prostate cancer via upregulating L-plastin. Cell Death Dis 8: e3060.
- Zheng J, Rudra-Ganguly N, Powell WC, Roy-Burman P (1999) Suppression of prostate carcinoma cell invasion by expression of antisense L-plastin gene Am J Pathol 155: 115-26.
- Siddiqui TA, Lively S, Vincent C, Schlichter LC (2012) Regulation of podosome formation, microglial migration and invasion by Ca(2+)-signaling molecules expressed in podosomes J Neuroinflammation 9: 250.
- El AK, Wiesner C, Linder S (2016) Metalloproteinase MT1-MMP islets act as memory devices for podosome reemergence. J Cell Biol 213: 109-25.
- Wiesner C, Faix J, Himmel M, Bentzien F, Linder S (2010) KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116: 1559-69.
- Gawden-Bone C, Zhou Z, King E, Prescott A, Watts C, et al. (2010) Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-142. J Cell Sci 123: 1427-37.
- Linder S, Wiesner C (2015) Tools of the trade: podosomes as multipurpose organelles of monocytic cells. Cell Mol Life Sci 72: 121-35.
- Linder S, Wiesner C (2016) Feel the force: Podosomes in mechanosensing. Exp Cell Res 343: 67-72.
- Cougoule C, Lastrucci C, Guiet R, Mascarau R, Meunier E, et al. (2018) Podosomes, But Not the Maturation Status, Determine the Protease-Dependent 3D Migration in Human Dendritic Cells. Front Immunol. 9: 846.
- Shinomiya H (2012) Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int J Cell Biol 2012: 213492.
- Chellaiah MA, Moorer MC, Majumdar S, Aljohani H, Morley SC, et al. (2020) L-Plastin deficiency produces increased trabecular bone due to attenuation of sealing ring formation and osteoclast dysfunction. Bone Res 8: 3.
- Chellaiah MA, Majumdar S, Aljohani H (2018) Peptidomimetic inhibitors of L-plastin reduce the resorptive activity of osteoclast but not the bone-forming activity of osteoblasts in vitro. PLoS ONE 13: e0204209.
- Majumdar S, Wadajkar AS, Aljohani H, Reynolds MA, Kim AJ, et al. (2019) Engineering of L-Plastin Peptide-Loaded Biodegradable Nanoparticles for Sustained Delivery and Suppression of Osteoclast Function In Vitro. Int J Cell Biol 2019: 6943986.
FIGURE 1
Figure 1: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE 2
Figure 2: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE 3
.jpg)
.JPG)
Figure 3: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE 4
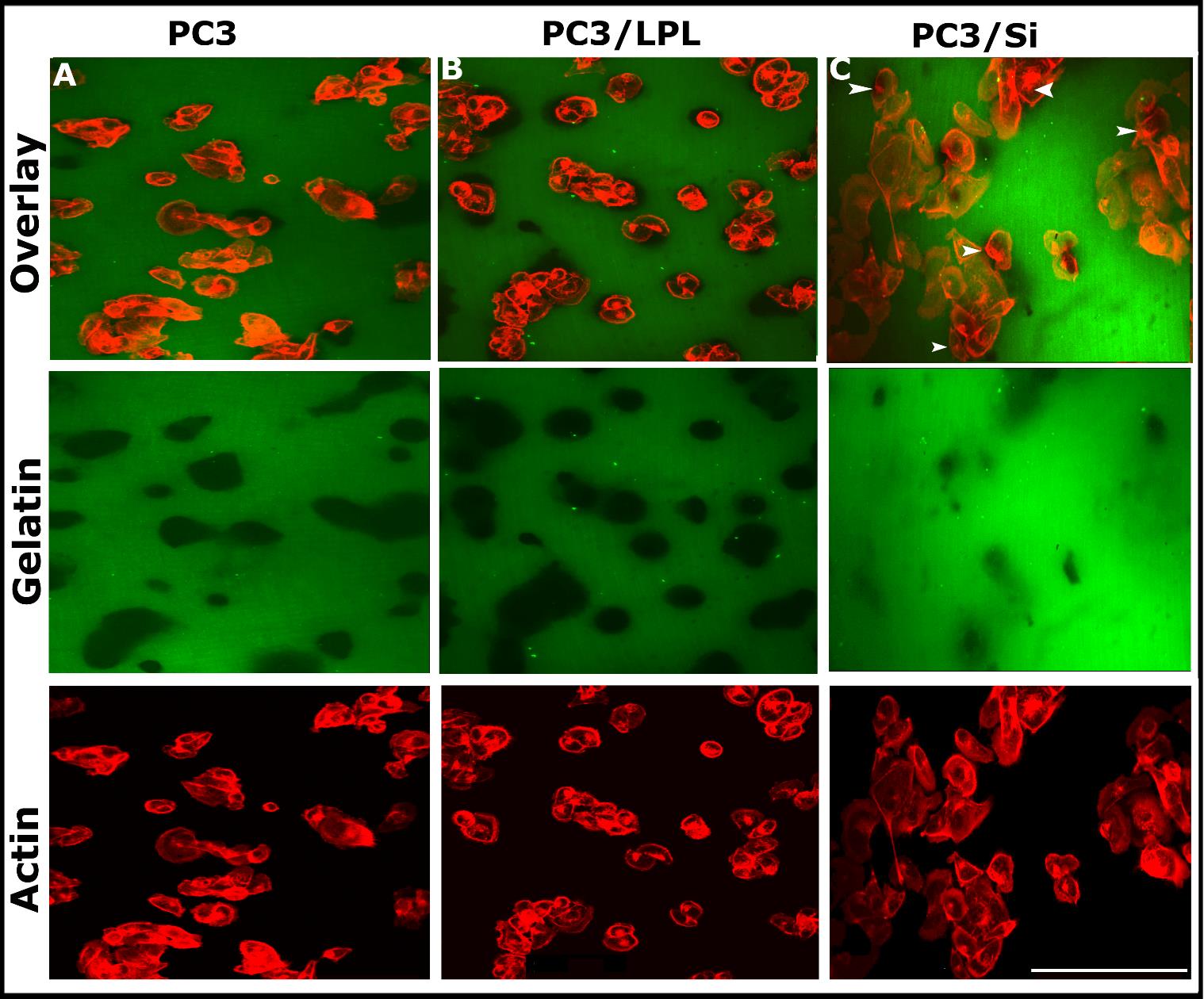
Figure 4: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE 5
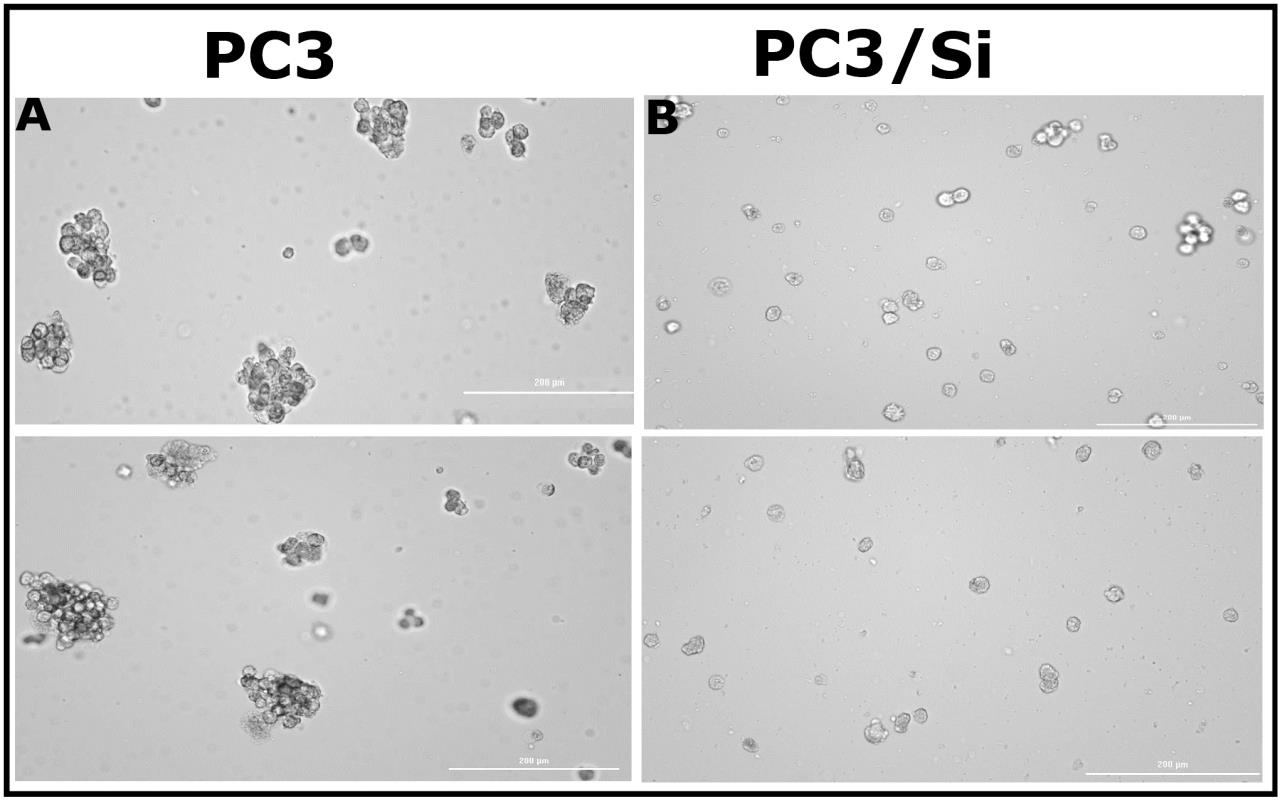
Figure 5: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE S1
Figure S1: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE S2
Figure S2: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE S3
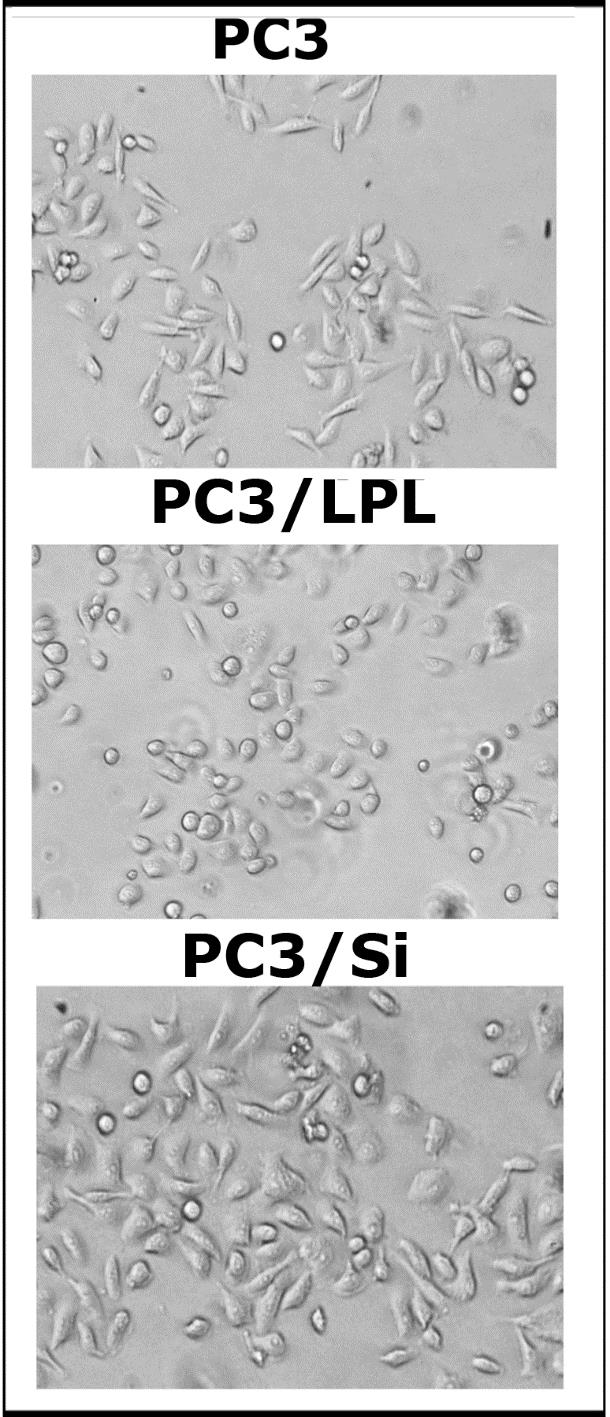
Figure S3: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE S4
Figure S4: Direct and indirect cost ($) by type of stay and surgery type (n=68)
FIGURE S5
Figure S5: Direct and indirect cost ($) by type of stay and surgery type (n=68)
Tables at a glance
Figures at a glance