Translationally Controlled Tumor Protein: A Key Target to Abrogate DNA Repair and Therapeutic Resistance in Cancer
Received Date: December 19, 2021 Accepted Date: January 19, 2022 Published Date: January 21, 2022
doi: 10.17303/jcrto.2022.10.101
Citation:Kenneth Omabe (2022) Translationally Controlled Tumor Protein: A Key Target to Abrogate DNA Repair and Therapeutic Resistance in Cancer. J Cancer Res Therap Oncol 10: 1-17.
Abstract
DNA damage response is a cellular survival mechanism exploited by cancer cells to exert resistance to therapies and has become a target of many antitumor agents. Translationally Controlled Tumor Protein (TCTP) is a multifunctional protein involved in malignant transformation, cancer progression and therapeutic resistance. Recently, several reports have incriminated TCTP in DNA repair mechanisms with new insights for explaining how TCTP partakes in this molecular activity. Here, we summarize and discuss DNA damage repair mechanisms in different contexts and highlight how TCTP manipulates the repair mechanism to drive cancer progression and chemoradiation resistance. Finally, we consider the future direction of this field, with a particular focus on combination strategies with TCTP inhibitors (i.e., gene inhibition, small molecules) and chemotoxicity to overcome TCTP-driven therapy resistance.
Keywords:TCTP; DNA Damage Response; HR; NHEJ; Chemoradiation; Cancer Therapy Resistance
DNA damage and repair mechanisms: a bird’s eye view
The human genome is constantly under threat from both external and internal sources. Environmental exposure to ionizing radiations or endogenously generated radical oxygen species (ROS) and replicative errors result in multiple forms of DNA lesions [1]. These lesions possess differing degrees of complexity ranging from, base damage, single strand breaks (SSB) to intra-and interstrand cross linkages (ICL), DNA-protein crosslinks (DPC), and double strand breaks (DSBs) [2] (Figure 1). How cells address DNA damage depends on the type of damage, cell cycle phase and availability of repair factors. Table 1 shows different types of DNA damage, their corresponding repair systems and activated genes in mammalian and yeast contexts (Table 1). In any case, when cells incur DNA damages, they activate checkpoint mechanisms and transcriptional signals that allows cell cycle arrest and lesion repair. If the damage is extensive and outweighs the cellular capacity to repair, cells will undergo cell death by apoptosis. If the damage is repaired, cells can then resume cycling, as part of the process known as checkpoint recovery [3]. If the damage is not repaired or incompletely resolved, cells can decide to override the checkpoint system and re-enter the cell cycle with damaged DNA and consequent genetic alterations, a process called checkpoint adaptation [3]. These genetic changes can be propagated to progeny, giving rise to mutations that promote the development of cancer and degenerative diseases (also called stochastic affects) [4]. These series of coordinated events through which cells manage genomic assaults are known as the DNA damage response (DDR) [5]. DDR is therefore a conglomeration of molecularly conserved DNA repair pathways consisting of distinct protein complexes that coordinate the maintenance of genomic integrity [5,6] (Table 1).
Eukaryotic cells possess extensive repair mechanisms to ensure the stability of the genome and cell survival, avoiding the consequences of propagating unrepaired or defectively repaired damage into progeny, which can lead to genomic instability and diverse pathological conditions such as cancer. While Single strand break (SSBs) and base damage lesions, which do not distort the helical structure of the DNA, are repaired by base excision repair (BER), bulky adducts and cross-linkages are repaired by nucleotide excision repair (NER) (Figure 1). Double strand breaks (DSBs) are resolved through a more complex repair mechanisms involving two separate intricate pathways referred to as Non-homologous end-joining (NHEJ) and Homologous recombination (HR) [7–11] and will constitute, largely the focus of our discussion. The DDR is coupled with apoptosis and cell cycle regulations such that, the cell cycle checkpoints kinases (CHK1 & CHK2) are signaled to halt or delay the cell cycle progression while repair is activated or, in a case of irreparable damage, the apoptotic signals are activated to execute the cells at the expense of genetic alteration [12]. Generally, maintaining the balance between apoptosis and proliferation during cellular response to DNA damage is of utmost importance in cellular homeostasis constitutes the major factors driving genome surveillance and maintenance [13–16].
Single strand breaks repair
DNA single-strand breaks (SSBs) are, most of the times, generated by cellular metabolic processes and/or external genotoxic agents. SSBs can cause double-strand breaks when they encounter replication folks in a repeated cell cycle. [17] Although SSB constitutes threats (less deleterious) to genomic integrity, they do not distort the helical structure of the DNA. However, several mechanisms have been described for SSB repair [7]. Most genotoxic agents such as ionizing radiation and some chemotherapies, upon exposure to DNA, generate differentforms of damage including lesions and double-strand breaks as well as SSBs.
Activation of poly (ADP-ribose) polymerase (PARP) is considered as a key primordial event that characterize cellular response to SSB. PARP binds to SSBs and uses NAD+ to polymerize PAR, thereby activating the recruitment of other relevant repair markers such as XRCC1 (X- Ray Repair Cross Complementing 1) to the site of damage [18]. XRCC1 reportedly binds to DNA ligase III and DNA polymerase β and interacts with BER repair effectors to execute repair. Inhibition of PARP blocks PARylation and recruitment of XRCC1 thereby generating DSB in a subsequent cycle. In cells harboring HR deficiency such as that found in a subset of familial breast and ovarian cancer patients due to BRCA1/2 germline or somatic mutation, synthetic lethality is created making PARP inhibitors effective anticancer drugs either as a monotherapy or in combination with DNA damaging agents [19, 20].
Translationally controlled Tumour Protein: one name, multiple functions
Translationally controlled Tumour Protein (TCTP, also named TPT1, p23 or fortilin) is a highly conserved multifunctional protein that is involved in regulating several biological processes. It is abundantly present in virtually all eukaryotic cells and possesses a high degree of homology between species with significant wide spectrum of distribution in several human tissues [23]. Since its discovery about three decades ago, TCTP has since received substantial functional and expressional characterization in both human and animal models. It is involved in cell growth and development, cell cycle regulation, cellular stress response, immune responses, apoptosis and autophagy [24–27]. TCTP is differentially expressed in a number of human cancers such as breast, ovarian and prostate cancers [28–30] where it, promotes cell migration, inversion and metastasis through induction of epithelial to mesenchymal Transition [31]. TCTP is a regulator of the cancer stem cell compartment [32] and has been identified as an important factor in tumour progression [33] and reversion [34,35]. Transient ectopic expression of TCTP protects both HeLa and U2OS cells from undergoing etoposide-induced apoptosis by the blockage of caspase-3-cleavage [36]. Moreover, TCTP was described as a pro- survival protein antagonizing BAX function [27]. Activation of P53 and Siah-1 downregulates TCTP expression both at protein and mRNA levels highlighting TCTP as a pro-tumour gene [37]. It is reportedly dysregulated in a few non-oncological diseases such as certain forms of inflammatory, metabolic and allergic diseases [38,39] and has since emerged a rational therapeutic target [40,41]. Data from our laboratory had shown that TCTP interactionwith hsp27: a stress-induced chaperone that is highly overexpressed in a number of cancer phenotypes including castration-resistant prostate cancer (CRPC), modulates the multifaceted functions of TCTP including it roles in DNA damage repair and RNA splicing [42]. Abrogation of this interaction with a Hsp27 inhibitor (OGX-427), restored sensitivity of CRPC to therapies [42]. TCTP is also reciprocally and negatively regulated by P53 [43]. TCTP competes with NUMB for MDM2 binding thereby interfering with p53-MDM2 complex formation and promoting MDM2-mediated ubiquitination and degradation of P53. Mice lacking TCTP are vulnerable to P53-dependent apoptosis while TCTP proficiency portends poor differentiation, tumor aggressiveness and poor prognosis in breast cancer phenotype [43].
An explicit involvement of TCTP in DNA repair recently emerged and has increasingly become a subject of attraction. This emerging role of TCTP has ostensibly gained enormous support due to the recently uncovered crosstalk between TCTP and key members of the DNA damage response (DDR) machinery such as ATM, DNA-PK, 53BP1 and p53 etc (Table 1) and has formed the basis for considering TCTP as a molecular player in the maintenance of genomic integrity [44–46]. Recently, a compilation of reports on the roles of TCTP in differentaspects of tumor biology including DNA damage and Repair was published suggesting, in general terms, the involvement of TCTP in genotoxic cellular response and possible therapeuticopportunities [47]. Although various views and perspectives have been presented in thiscontext, the specific nature of DDR-TCTP interface has not been fully defined. In this review,we provide advanced knowledge of DNA repair mechanisms with particular reference to NHEJ and HR signal transductions that culminate to DNA repair. We bring an insight into how TCTP-dependent regulation of the key recombination markers at different stages of the DNA repair signal transduction, affects cellular behaviour and response to DNA damage induction. We also elaborate on the therapeutic possibilities associated with the DDR-TCTP intersection and suggest further approaches to better address unresolved issues bordering on TCTP-DNA repair interplay.
TCTP Roles in damage sensing and mediation
Upon DNA damage induction, the sensor factors detect the damage signals, identify and locate the damage sites. The damage signals are amplified to the transducers or mediators, which in turn, relay the signal to the effector complex of proteins [48]. Damage sensing and recognition is an important aspect of DNA repair signaling which allows cells to recognize the presence of damage and set in motion the damage recovery system. The MRN complex (Mre-11-Rad50- Nbs1) activation is the major early sensory indicator of DSB which further activates ATM or ATR depending on the source of damage [49–51]. MRN-dependent activation of ATM induces ATM autophosphorylation which in turn, phosphorylates H2AX at ser139 to form yH2AX as well as CHK2 which can all be visualized as foci at the damage site [52] (Figure 2a). CHK2 phosphorylates p53, which acts as a transcription factors to other repair factors as well as causes cell cycle inhibition through p21[53]. Contrary to ATM that is activated by IR-induced DSB, ATR is activated by internally generated DSB such as replication fork stalling and oxidative stress-induced DSBs [50]. Both ATM and ATR phosphorylate H2AX to amplify the damage signal and recruit repair mediators to the damage site for other downstream events [50].
There is compelling evidence that TCTP is required for the damage sensing signaling and may be indispensable in this regard. Using normal human fibroblast, Zhang et al. demonstrated that TCTP is upregulated following cellular exposure to low dose (10Gy delivered at 0.2Gy/h) of gamma-radiation, translocates to the nucleus and exists in complex with some critical factors of DNA damage signal sensing and transductions such as ATM, yH2AX, 53BP1 and Ku70/80 [44]. Upregulation of TCTP was in a manner dependent on ATM and depletion of TCTP resulted in a decrease in the DNA binding ability and nuclear abundance of Ku70/80, attenuation of ATM kinase activity as well as delayed YH2AX foci formation. Consequently, cells that lacked TCTP failed to recover from irradiation-induced chromosomal damage [44].
Similarly, an independent in-vivo experiment investigating the role of TCTP in growth regulation in Drosophila models, demonstrated that TCTP (dTCTP) directly modulates the activity of Drosophila ATM (dATM) and mutation of dTCTP resulted in a significant increase to radiation sensitivity, defects in growth rate and chromosomal stability [54]. Altogether, these suggest that TCTP is required for the recruitment of ku70/80 and ATM respectively to the site of damage and plays important roles in the upstream events of both NHEJ and HR pathways. Therefore, it is attractive to infer that TCTP is indispensable in damage sensing and mediation.
TCTP roles in Non-Homologous End Joining (NHEJ)
Non-Homologous End Joining (NHEJ) is the error prone repair pathway of DSBs occurring predominantly at the G1 phase of the cell cycle. Ku70/80 is the DNA-binding subunit of DNA- dependent protein kinase (DNA-PK) and an exclusive marker of NHEJ. TCTP association with the KU-complex has been reported and highlight the role of TCTP in regulating the NHEJ via the KU-complex [55]. Upon cellular decision to commit repair through the NHEJ, Ku70/Ku80 heterodimer senses the signal and recruits other critical components of the repair pathway including p53-binding protein 1 (53BP1), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), XRCC4, ligase IV, XLF and Artemis (Figure 1), which then process the broken DNA and seal the two ends [8,56]. The DNA-PK phosphorylates the histone H2AX that in turn recruits other effector proteins to the damaged site for onward execution of repair. While 53BP1 acts to protect the DSB ends from undergoing end resection, DNA-PK stabilizes the DSB end through the phosphorylation of Artemis. Artemis facilitates the end processing suchthat a complex of protein factors such as DNA Ligase 4 (LIG4), X-ray repair cross- complementing protein 4 (XRCC4), XLF-XRCC4-like factor, interact to complete the repair process. Due to the special end processing requirement for the generation of ligatable DNA ends associated with radiation-induced DSB, this repair pathway is usually characterized by base pairs deletions, random exchange of nucleotides and generalized sequence alteration [57].
TCTP involvement in the NHEJ pathway is evidenced in its interaction with the Ku- complex (ku70/80). TCTP depletion attenuates the nuclear abundance of the ku70/80 complex upon irradiation of normal human cells, implying that TCTP is required for both nuclear translocation of ku70/ku80 and other ku-related nuclear functions. Accordingly, studies have shown that protein translocation across biological membranes requires molecular chaperons to manage their loosely folded structures for efficient transmembrane transport [58,59]. This supports the assertion that TCTP exhibits its chaperon-like activity [60] on the ku70/80 thereby facilitating its nuclear translocation and function. In addition, since TCTP is found in complex with the ku70/80 at the site of damage, it further suggests that TCTP may play an important function, alongside the ku-complex, in the recruitment process driving NHEJ activation. Moreover, Ku-binding activation predominantly occurs at the G1 phase of the cell cycle. Independently, knockdown of TCTP has been shown to induce cell cycle arrest and prevent G1/S-phase transition under normal condition [61,62]. Put together, this shows that TCTP interaction with the ku promotes other NHEJ-dependent downstream events relating to cell cycle regulation. As opposed to its functions in an unstressed state, we propose that TCTP is involved in functions relating to checkpoint adaptation, damage bypass, and attenuation of cell cycle regulators and restarting of the cell cycling process under stressed condition. It is pertinent to note that NHEJ exhibits fast, quick and error-prone repair kinetics consequent upon possibilities of genetic alterations [8]. Why and how NHEJ pathway exhibits this peculiar characteristic is not entirely clear. To this end, we assert that the error-prone designate associated with NHEJ may be connected, at least in part, to the TCTP functions in NHEJ downplaying the effective DNA repair processes and apoptosis in favour of proliferation and cell survival. Overall, this goes further to connote that ‘while ideal DDR signals promote cell death at the expense of mutated cell life; arising from unrepaired/defectively repaired DNA damage, TCTP may promote genetically aberrated cell life at the expense of cell death’ and may explain the involvement of TCTP in cancer progression and therapy resistance asdiscussed later in this article. Secondly, it also suggests that TCTP cellular functions occur in a context- dependent manner. However, TCTP functions in NHEJ require further experimental validations.
TCTP roles in Homologous recombination (HR)
In response to DSB, damage recognition by MRN complex and ATM activation characterize the early events of the HR pathway [49]. One of the critical steps of the HR repair is the DNA nucleolytic end resection that is orchestrated by MRE11 mediated by CtIP and EXO1 (Exonuclease1) in the presence of BRCA1 and BLM (Bloom’s syndrome helicase). Contrary to the NHEJ, the HR repair pathway has a slower kinetics and requires a homology sequence from the sister chromatid to achieve error-free repair. While NHEJ occurs in almost all phases of the cell cycle, HR repair is confined at the S/G1-M phases where a more accurate repair is of the greatest essence [22,63] (Figure 2a).
A study investigating TCTP partner proteins in HeLa cells revealed that several HR markers are significantly enriched in the TCTP potential interactomes [45]. First in the list of proteins identified are BRATI and BCCIP, which are binding partners of BRCA1 and BRCA2 respectively. In addition to BRCA related proteins, MREII and NBSI components of the MRN complex were also found in the interactomes as well as ATR and Rad51b [45]. Further investigations reveal that TCTP associates with Rad51 and this association results in the stabilization of Rad51 at the damage sites. Inhibition of TCTP decreases the stability of Rad51 and impaired HR efficiency in MCF7. Furthermore, sertraline-dependent inactivation of TCTP promotes apoptosis induced DNA damage and resensitized MCF-7 to etoposide and Olaparib [45]. Interaction with MRN corroborates our earlier thought that TCTP may play an important function in damage sensing and mediation upstream of ATM activation. Be it as it may, TCTP functions in HR remain an open subject of investigation and discussion. From a second point of view, we recommend an investigation to ascertain whether TCTP mutation can be associated with cellular susceptibility to carcinogenesis, which will also inform if the contribution of TCTP to HR repair as reported is sufficient to distort HR operation upon TCTP depletion. Conversely, cancer cells adopt HR repair route to launch resistance against chemotherapies, especially at delicate cell cycle phases prior to replication [64], it is important to investigate whether the previously reported role of TCTP in chemo and radio resistance [33,65] is associated with enhanced DNA repair through the HR, and questions whether TCTP is a cancer susceptibility gene?
Polo-like kinase-1 (PLK1) and TCTP phosphorylation during DNA repair
Cellular checkpoint mechanisms are activated following DNA damage induction. This results to cell cycle arrest which allows cells to either repair damage or be committed to apoptosis. Conversely, for reasons not clearly understood, cells could decide to adapt the damage, re-enter the cell cycle and progress with replication at the expense of death in a process called checkpoint adaptation [66]. One of the key targets of the DNA damage checkpoint is Polo-like Kinase-1 (PLKI) and has recently emerged a drug target in cancer [67]. PLKI is involved in DNA damage repair through regulation of Cdk1 required to restart the cell cycle following a DNA damage-induced arrest [68]. TCTP is a substrate of PLK1 in a mechanism driving cell cycle progression. PLKI phosphorylates TCTP at ser46 residue to allow mitotic spindle segregation enabling mitosis completion and cytokinesis. Phosphorylation of TCTP correlates well with PLK1 level and kinase activity in cells [69]. Moreover, PLK1 depletion by siRNA or inactivation by specific inhibitors caused a corresponding decrease in phospho-TCTP-Ser46 signal validating TCTP activation as a direct marker of PLK1 activity [69]. TCTP is involved in tubulin binding and microtubule stabilization during the cell cycle. At metaphase, TCTP binds to the mitotic spindle and is subsequently detached from the spindle during the metaphase-anaphase transition [64], which is driven by TCTP phosphorylation by PLKI. PLKI activity was decreased by TCTP knockdown [69]. In normal conditions, when cells in G2 phase are challenged with DNA damage, several key mitotic regulators such as PLK1 are inhibited to prevent entry into mitosis [70]. This inhibition blocks the phosphorylation of TCTP thereby blocking cell cycle progression. However, studies have shown that expression of PLK1 is associated with TP53 inactivation, DNA repair deficiency, and cancer progression [71]. Furthermore, inhibition of PLK1 sensitizes cancer cells to radiotherapy in a manner dependent on p53 status [72]. Put together, this provides a link between DNA repair signaling, p53, TCTP, PLK1 and cancer resistance. Our understanding of this molecular interplay suggests that, upon DNA damage induction especially at G2/M-phase, expression of PLKI is inhibited which in turn blocks TCTP phosphorylation and cell cycle progression to allow time for repair.However, in a feedback loop mechanism, TCTP activates p53 degradation and orchestrates anostensible repair deficiency thereby re-inducing PLK1 expression and cell cycling process. By this explanation, TCTP downplays the effective repair process by negatively regulating p53, encouraging defective repair, adaptive mutability and cancer resistance.
TCTP and P53 pathway: a peep through the lens of DNA repair mechanisms
DNA damage downstream events are executed by the effector proteins: a component of DDR signaling that is involved in halting the cell cycle, apoptosis activation and DNA recovery processes. The key players of these DNA repair downstream events include P53, CHKI, CHK2CDC25A and CDC25C amongst others. CHK1 and CHK2 are considered targets of ATR and ATM respectively as a response mechanism to IR-induced and replication-induced DSBs respectively [73] (Figure 2b). Among these downstream factors, TCTP reportedly interacts with p53 in a manner that defines reciprocal inhibition [43]. The p53 is a tumour suppressor protein that plays a central role in determination of cell fate whether to undergo cell cycle arrest or apoptosis following different types of cellular stress such as hypoxia, oncogene activation or DNA damage [74]. P53 levels are generally low in normal cells due to Mdm2 mediated ubiquitination and degradation through the proteasome pathway or through facilitation of nuclear export [75]. As indicated above, upon DNA damage, ATM and ATR phosphorylate p53 at several sites in its trans-activation domain, including at Ser15 and Ser20 residues [76]. This phosphorylation inhibits the interaction of p53 with Mdm2 resulting in p53 stabilization [77]. In addition, ATM phosphorylates Mdm2 and decreases its ability to promote nucleo– cytoplasmic shuttling and the subsequent degradation of p53 [78]. This enhances p53 downstream signaling leading to cell cycle arrest and apoptosis activation (Figure 2b). How TCTP plays a role in P53-dependent response to DNA damage has not been clearly elucidated previously even though TCTP direct interaction with MDM2 and P53 is evident in several studies [33,43,79]. However, we consider three factors in this regard: (1) TCTP possesses anti- apoptotic activity through regulation of Bcl-2, Bcl-Xl, and Mcl-1 [27] as opposed to p53. (2) TCTP promotes cell cycle progression [26] as opposed to p53 during stress. (3) p53 and TCTP antagonize each other via separate routes (promoter regulation and MDM2-mediation respectively) [43]. Put together, it is persuasively convincing that the pro-survival properties of TCTP are not unconnected to its antagonistic action on P53. Whereas p53 promotes cell cycle arrest, DNA repair where repair is feasible and apoptosis or senescence where otherwise (Figure 3), TCTP promotes cell cycle progression whether or not the damage is resolved in favour of proliferation.
In oncological context, one way TCTP confers therapy resistance on cancers is through its inhibitory activity on P53 via the stabilization of the E3 ubiquitin Ligase (MDM2) and p53 proteasome-dependent degradation [80]. From these interactions, it is reasonable to propose that the anti-p53 activity of TCTP promotes DNA repair without recourse to the consequences of repair defects arising from genetic alterations. This is also supported by the fact that TCTP promotes NHEJ repair, which is an error-prone pathway, through facilitation of K70/80 binding to the DNA and activation of NHEJ downstream signaling [44]. This kind of cellular behavioris famous in cancer cells proliferation and resistance to DNA damaging agents. This may explain why TCTP has been consistently implicated in cancer progression and resistance to therapies [24,33]. As a result, combination of TCTP inhibition with chemotherapy is a promising treatment strategy targeting cancer cells. However, it is recommended that investigations that are more detailed, be performed to unravel how TCTP drives cellular recovery from DSBs overriding signal contributions for cell cycle arrest, apoptosis andsenescence
Implications for cancer progression and therapy resistance
TCTP is overexpressed in different cancer types such as breast, ovarian, liver, lungs, skin, colon and prostate [29,30,33,81]. Studies have shown that overexpression of TCTP correlates well with the tumor progression and resistance to therapies. For example, our protracted works on TCTP and prostate cancer had previously established a strong association between TCTP and prostate cancer progression and resistance to both hormonal and chemical drugs. Inhibition of TCTP using ASO-strategy resensitized PC cells to docetaxel treatment [40].
In another experiment, TCTP protects A549 cells from irradiation induced DNA damage while depletion of TCTP exposes the cells to irradiation-induced cell death [65]. Similarly, Oxaliplatin and 5-FU-induced DNA damage in colorectal cancer cells resulted in a 4-fold increase in TCTP protein level and protection from the consequences of irradiation. Conversely, TCTP knockdown sensitized HCT116 to 5-FU and Oxaliplatin–induced cytotoxicity [61]. These findings indicate that TCTP is indispensable in cancer progression, aggressiveness and resistance to chemotherapies. However, no mechanism has been clearly elucidated abintio, to underlie this indication. Here, we put forward a hypothesis that TCTP involvement in DNA repair may underlie the exacerbation and resistance properties observed in these cancer types with proficient expression of TCTP. As partly described above, while we support the notion that TCTP promotes DNA repair notably through the NHEJ pathway, this promotion may only be to a limited extent capable to prevent apoptosis and drive proliferation in disease context. This proposal is supported by the fact that, while DDR signals promote apoptosis or DNA repair (depending on the nature of damage and micro environmental factors) upon cell cycle arrest [82], TCTP inhibits apoptosis and promotes cell cycle progression and DNA repair [27]. In other words, TCTP downplays the signals for effective DNA repair, thereby promoting adaptive mutability in cancer. This confers a more complex and aberrated genetic architecture to cancers allowing them to evade cytotoxicity and engage in accelerated proliferation. From this point of view, we can infer that TCTP is a proliferation bias protein involved in driving cancer progression and resistance to therapy through adaptive mutability during DNA repair. We further describe below, an intervention strategy that can complement the effect of therapeutic agents in cancer treatment.
Combinational therapy
Inhibition of TCTP in combination with a DNA damaging agent potentially provides a greatly promising intervention strategy to combat TCTP-dependent cancer resistance. This could be achieved by using TCTP inhibitors such as TCTP-targeting Antisense Oligonucleotides (ASOs), Sertraline, Thioridazine (Figure 4) in parallel with or prior to the use of DNA poisons. Alternatively, we propose the inhibition of TCTP expression in combination with a DNA damaging agent combined in a single nano formulation. These therapeutic combinatorial approaches will synergistically combine the effects of gene inhibition and cytotoxicity to achieve enhanced efficacy. To inhibit TCTP at the mRNA level, we have previously developed a Lipid-conjugated Antisense Oligonucleotide (LASO) therapy for targeting TCTP. LASO is able to self-assemble into small particles, organized into nanomicelles in an aqueous media offering a micellar core that can encapsulate antitumor agents for enhanced efficacy (Figure 4). Inhibition of TCTP using Lipid-ASO (LASO) correlated well with tumor sensitivity to cytotoxic drugs such as paclitaxel [41].
Similarly, studies have shown that micelles represent a candidate vehicle for both carriage and solubilisation of antitumor agents. In preclinical models of prostate cancer for example, combination of docetaxel (antimitotic agent), rapamycin (mTOR inhibitor) and 17-N- allylamino-17-demethoxygeldanamycin (HSP90 inhibitor) in a single micellar system resulted in a more efficient inhibition of tumor growth in vitro compared to their individual efficacy and cytotoxic effects of the drugs were more effective with micellar –dependent delivery [83].
In this present study, TCTP is highlighted as a driver of adaptive mutability through defective DNA damage repair and blockage of apoptosis, thereby promoting replicative immortality in cancer cells and resistance to therapy. Our proposed chemogene conjugate (consisting of TCTP-gene inhibitor and antitumor agents encapsulated by micelle) takes advantage of its amphiphilic property and forms an attractive strategy for synergising gene inhibition and cytotoxicity in a combinatorial strategic cancer therapy. In addition, this approach will enhance delivery of chemotherapeutics with decreased rate of non-specific cytotoxicity.
Concluding remarks
TCTP plays important role in DNA damage repair. This study reveals different aspects of DNA repair mechanisms where TCTP is involved. First, we discussed cellular mechanisms of DNA damage repair in different contexts. We reviewed several reports linking TCTP and DNA damage response. Based on the available knowledge and scientific evidence,we provide a novel insight that clearly elucidates the role of TCTP in DNA damage repair. We conclude that TCTP promotes DNA repair in a manner that downplays the effective repair mechanism in favours of proliferation and cell cycle progression thereby facilitating adaptive mutability leading to mutagenesis and therapy resistance in cancer. We then propose a combinatorial therapeutic strategy that will harbour TCTP-targeting ASO as TCTP inhibitor and DNA damaging agent in a single nanoformulation to combat TCTP-driven therapy resistance. Further studies on underlying mechanisms for the roles of TCTP in DDRs are recommended.
Author Contributions
KO, PR and DT conceived and designed the work. KO, TKL, DT and CP wrote the manuscript. All authors analyzed and critically read the manuscript before submission.
Funding
The authors would like to thank ITMO cancer PSCI (Grant Number #C21033AS) for the funding of this work. We are also grateful to INSERM, AEFUNAI, TETFund Nigeria, Campus France and Amidex Foundation for their support.
Conflicts of Interest
The authors declare no conflict of interest.
- Cadet J, Richard Wagner J (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol 2013: 5.
- Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461: 1071-8.
- Zhou BBS, Elledge SJ (2000) The DNA damage response: Putting checkpoints in perspective. Nature 408: 433-9.
- Basu AK (2018) DNA damage, mutagenesis and cancer. Int J Mol Sci 2018: 19.
- Ciccia A, Elledge SJ (2010) The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 40: 179-204.
- Niida H, Nakanishi M (2006) DNA damage checkpoints in mammals. Mutagenesis 21: 3-9.
- Caldecott KW (2008) Single-strand break repair and genetic disease Nat Rev Genet 9: 619-31.
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18: 495-506.
- McKinnon PJ, Caldecott KW (2007) DNA strand break repair and human genetic disease. Annu Rev Genomics Hum. Genet 8: 37-55.
- Kavanagh JN, Redmond KM, Schettino G, Prise KM (2013) DNA double strand break repair: A radiation perspective. Antioxidants Redox Signal 18: 2458-72.
- Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39-85.
- Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med 12: 440–50.
- Vindigni A (2017) Special issue: “At the Intersection of DNA Replication and Genome Maintenance: from Mechanisms to Therapy.” Biophys. Chem 225: 1-2.
- Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208-19.
- Toueille M, Hübscher U (2004) Regulation of the DNA replication fork: A way to fight genomic instability. Chromosoma 113: 113-25.
- Toledo L, Neelsen KJ, Lukas J (2017) Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 66: 735-49.
- Demin AA, Hirota K, Tsuda M, Adamowicz M, Hailstone R, et al. (2021) XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 81: 3018-30.
- The role of PARP-1 in the repair of single stranded break (SSB) Antibody News: Novus Biologicals.
- Nizialek E, Antonarakis ES (2020) PARP Inhibitors in Metastatic Prostate Cancer: Evidence to Date. Cancer Manag Res 2020, 12: 8105.
- W A, D C, A P, JD S, B S, et al. (2020) Non- BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res 26: 2487-96.
- Polo SE, Jackson SP (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25: 409.
- Lozano R, Castro E, Aragón IM, Cendón Y, Cattrini C, et al. (2021) Genetic aberrations in DNA repair pathways: a cornerstone of precision oncology in prostate cancer. Br J Cancer 124: 552-63.
- Bommer UA, Thiele BJ (2004) The translationally controlled tumour protein (TCTP). Int J Biochem Cell Biol 36: 379-85.
- Koziol MJ, Gurdon JB (2012) TCTP in development and cancer. Biochem. Res Int.
- Bommer UA (2017) The translational controlled tumour protein TCTP: Biological functions and regulation. In Results and Problems in Cell Differentiation; Springer Verlag 64: 69-126.
- Jojic B, Amodeo S, Ochsenreiter T (2018) The translationally controlled tumor protein TCTP is involved in cell cycle progression and heat stress response in the bloodstream form of Trypanosoma brucei. Microb Cell 5: 460-9.
- Susini L, Besse S, Duflaut D, Lespagnol A, Beekman, C, et al. (2008) TCTP protects from apoptotic cell death by antagonizing bax function. Cell Death Differ 15: 1211-20.
- Baylot V, Karaki S, Rocchi P (2017) TCTP Has a Crucial Role in the Different Stages of Prostate Cancer Malignant Progression. In TCTP/tpt1 - Remodeling Signaling from Stem Cell to Disease; Telerman, A., Amson, R., Eds.; Results and Problems in Cell Differentiation; Springer International Publishing: Cham 2017: 255-261.
- Li S jie, Jia H yao, Wu D, Fan Z (2011) min Nan Fang Yi Ke Da Xue Xue Bao 31: 1560-3.
- Chen C, Deng Y, Hua M, Xi Q, Liu R, et al. (2015) Expression and clinical role of TCTP in epithelial ovarian cancer. J. Mol. Histol 46: 145-56.
- Bae SY, Kim HJ, Lee KJ, Lee K (2015) Translationally controlled tumor protein induces epithelial to mesenchymal transition and promotes cell migration, invasion and metastasis. Sci Rep 5: 8061.
- Amson R, Pece S, Marine JC, Fiore PP (2013) Di; Telerman, A. TPT1/ TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol 23: 37-46.
- Baylot V, Katsogiannou M, Andrieu C, Taieb D, Acunzo J, et al. (2012) Targeting TCTP as a new therapeutic strategy in castration-resistant prostate cancer. Mol. Ther. J. Am. Soc. Gene Ther 20: 2244-56.
- Tuynder M, Susini L, Prieur S, Besse S, Fiucci G et al. (2002) Biological models and genes of tumor reversion: Cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci USA 99: 14976-981
- Tuynder M, Fiucci G, Prieur S, Lespagnol A, Géant A, et al. (2004) Translationally controlled tumor protein is a target of tumor reversion. Proc. Natl. Acad. Sci. USA 101: 15364-69.
- Li F, Zhang D, Fujise K (2001) Characterization of Fortilin, a Novel Antiapoptotic Protein. J Biol Chem 276: 47542-549.
- Telerman A, Amson R (2009) The molecular programme of tumour reversion: The steps beyond malignant transformation. Nat. Rev. Cancer 9: 206-16.
- Bommer UA, Telerman A (2020) Dysregulation of TCTP in Biological Processes and Diseases. Cells 2020: 9.
- Kang HS, Lee MJ, Song H, Han SH, Kim YM et al. (2001) Molecular Identification of IgE-Dependent Histamine-Releasing Factor as a B Cell Growth Factor. J Immunol 166: 6545-54.
- Acunzo J, Baylot V, So A, Rocchi P (2014) TCTP as therapeutic target in cancers. Cancer Treat Rev 40: 760-9.
- Karaki S, Benizri S, Mejías R, Baylot V, Branger N, et al. (2017) Lipid-oligonucleotide conjugates improve cellular uptake and efficiency of TCTP-antisense in castration-resistant prostate cancer. J Control Release 258: 1-9.
- Katsogiannou M, Andrieu C, Baylot V, Baudot A, Dusetti NJ, et al. (2014) The functional landscape of Hsp27 reveals new cellular processes such as DNA repair and alternative splicing and proposes novel anticancer targets. Mol Cell Proteomics 13: 3585-601.
- Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, et al. (2012) Reciprocal repression between P53 and TCTP Nat Med 18: 91-9.
- Zhang J, De Toledo SM, Pandey BN, Guo G, Pain D, et al. (2012) Role of the translationally controlled tumor protein in DNA damage sensing and repair. Proc Natl Acad Sci USA 2012: 109
- Li Y, Sun H, Zhang C, Liu J, Zhang H, et al. (2017) Identification of translationally controlled tumor protein in promotion of DNA homologous recombination repair in cancer cells by affinity proteomics. Oncogene 36: 6839-49.
- Li S, Chen M, Xiong Q, Zhang J, Cui, Z, et al. (2016) Characterization of the Translationally Controlled Tumor Protein (TCTP) Interactome Reveals Novel Binding Partners in Human Cancer Cells. J Proteome Res 15: 3741-51.
- Li S, Ge F (2017) TCTP/tpt1 - Remodeling Signaling from Stem Cell to Disease 64: 127-36.
- Lieber MR (2008) The mechanism of human nonhomologous DNA End joining. J Biol Chem 283: 1-5.
- Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Sci 308: 551-4.
- ATR disruption leads to chromosomal fragmentation and early embryonic lethality.
- Syed A, Tainer JA (2018) The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem 87: 263-94.
- Jackson SP (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis 23: 687-96.
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, et al. (1995) p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev 9: 935-44.
- Hong ST, Choi KW (2013) TCTP directly regulates ATM activity to control genome stability and organ development in Drosophila melanogaster. Nat Commun 2013: 4.
- Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, et al. (2008) Funabiki, H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol 182: 467-79.
- Yang K, Guo R, Xu D (2016) Non-homologous end joining: Advances and frontiers. Acta Biochim. Biophys. Sin. (Shanghai) 48: 632-40.
- Harper JW, Elledge SJ (2007) The DNA Damage Response: Ten Years After. Mol. Cell 28: 739-45.
- Craig EA (2018) Hsp70 at the membrane: Driving protein translocation. BMC Biol 2018: 16.
- Dierks T, Klappa P, Wiech H, Zimmermann R (1993) The role of molecular chaperones in protein transport into the endoplasmic reticulum. Philos. Trans. R. Soc. Lond. B Biol Sci 339: 335-41.
- Chan THM, Chen L, Guan XY (2012) Role of translationally controlled tumor protein in cancer progression. Biochem Res Int.
- Bommer UA, Vine KL, Puri P, Engel M, Belfiore L, et al. (2017) Translationally controlled tumour protein TCTP is induced early in human colorectal tumours and contributes to the resistance of HCT116 colon cancer cells to 5-FU and oxaliplatin. Cell Commun. Signal 15: 1-15.
- Wang L, Tang Y, Zhao M, Mao S, Wu L, et al. (2018) Knockdown of translationally controlled tumor protein inhibits growth, migration and invasion of lung cancer cells. Life Sci 193: 292-9.
- Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58: 235-63.
- Jeon HJ, You SY, Park YS, Chang JW, Kim JS, et al. (2016) TCTP regulates spindle microtubule dynamics by stabilizing polar microtubules during mouse oocyte meiosis. Biochim. Biophys. Acta - Mol. Cell Res 1863: 630-7.
- Jung J, Lee JS, Lee YS, Lee K (2019) Radiosensitivity of cancer cells is regulated by translationally controlled tumor protein. Cancers (Basel) 2019: 11.
- Bahassi EM (2011) Polo-like kinases and DNA damage checkpoint: Beyond the traditional mitotic functions. Exp Biol Med 236: 648-57.
- Degenhardt Y, Lampkin T (2010) Targeting polo-like kinase in cancer therapy. Clin. Cancer Res 16: 384-9.
- Hyun SY, Hwan HI, Jang YJ (2014) Polo-like kinase-1 in DNA damage response. BMB Rep 47: 249-255.
- Cucchi U, Gianellini LM, De Ponti A, Sola F, Alzani R, et al. (2010) Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res 30: 4973-86.
- Bruinsma W, Aprelia M, Garciá-Santisteban I, Kool J, Xu YJ, et al. (2017) Inhibition of Polo-like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene 2017, 36: 1840-48.
- High expression of polo-like kinase 1 is associated with TP53 inactivation, DNA repair deficiency, and worse prognosis in ER positive Her2 negative breast cancer.
- Van den Bossche J, Domen A, Peeters M, Deben C, De Pauw I, et al. (2019) Radiosensitization of non-small cell lung cancer cells by the plk1 inhibitor volasertib is dependent on the p53 status. Cancers (Basel) 2019: 11.
- Zannini L, Delia D, Buscemi G (2014) CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 6: 442-57.
- Williams AB, Schumacher B (2016) p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med 2016: 6.
- Carr MI, Jones SN (2016) Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res 5: 707-24.
- Bar J, Grelewski P, Deszcz I, Noga L, Hirnle L, et al. (2018) Association between p53 protein phosphorylated at serine 20 expression and ovarian carcinoma stem cells phenotype: correlation with clinicopathological parameters of ovarian cancer. Neoplasma 66: 801-9.
- Caspari T (2000) Checkpoints: How to activate p53. Curr Biol 2000: 10.
- Gannon HS, Woda BA, Jones SN (2012) ATM Phosphorylation of Mdm2 Ser394 Regulates the Amplitude and Duration of the DNA Damage Response in Mice. Cancer Cell 21: 668-79.
- Funston G, Goh W, Wei SJ, Tng QS, Brown C, et al. (2012) Binding of translationally controlled tumour protein to the N- terminal domain of HDM2 is inhibited by Nutlin-3. PLoS One 2012: 7.
- Boia-Ferreira M, Basílio AB, Hamasaki AE, Matsubara FH, Appel MH, et al. (2017) TCTP as a therapeutic target in melanoma treatment. Br. J. Cancer 117: 656-65.
- Du J, Yang P, Kong F, Liu H (2017) Aberrant expression of translationally controlled tumor protein (TCTP) can lead to radioactive susceptibility and chemosensitivity in lung cancer cells. Oncotarget 8: 101922-35.
- Malaquin N, Carrier-Leclerc A, Dessureault M, Rodier F (2015) DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015: 5.
- Le B, Powers GL, Tam YT, Schumacher N, Malinowski RL, et al. (2017) Multi-drug loaded micelles delivering chemotherapy and targeted therapies directed against HSP90 and the PI3K/AKT/mTOR pathway in prostate cancer. PLoS One 12: e0174658.
FIGURE 1
Single strand break (SSBs) and base damage lesions which do not distort the helical structure of the DNA are repaired by base excision repair (BER), bulky adducts and cross-linkages are repaired by nucleotide excision repair (NER) (See figure 1 for DNA damage types and appropriate repair routes). Double strand breaks (DSBs) are resolved through a more complex repair mechanisms involving two separate intricate pathways referred to as Non-homologous end-joining (NHEJ) and Homologous recombination (HR)
Figure 1: Forms of DNA damage, their corresponding repair pathways and signalling complexes
FIGURE 2
.JPG)
TCTP interacts with the component of the sensor complex comprising the ku70/80 complex, MRN and ATM resulting in the Phosphorylation of yH2AX that amplifies the damage and recruitment process. Binding of ku-80/70 and NHEJ activation is predominant at the G1-early S-phase of the cell cycle. Mediating proteins (53BP1 and BRCA1) compete for binding to the damage site to prevent or promote DNA end resection respectively, which determines the repair pathway of choice. Binding of BRCA1 and activation HR is predominant at late S-phase/G2 cell cycle phas
Figure 2(a): A schematic illustration of TCTP involvement in DSB repair signaling through the NHEJ and HR pathways
.JPG)
Execution of downstream events such as cell cycle arrest, apoptosis activation, and DNA recovery by the effector protein. ATM activates CHK2 which further activates p53, CDC25C, CDC25A to generally halt the cell cycle, activate apoptosis
Figure 2(b): A schematic illustration of TCTP involvement in downstream DSB repair signaling through the NHEJ and HR pathways
FIGURE 3
p53 plays a central role in DNA damage responses (DDRs), which could lead to cell cycle arrest, DNA repair, senescence or apoptosis upon DNA damage induction. DNA damage activates sensor proteins such as ATM and ATR (1). Activated ATM then phosphorylates CHK2 (2) that subsequently is responsible for phosphorylation of p53 (3). Similarity, activated ATR phosphorylates CHK1 (4) that is required for p53 phosphorylation (5). In addition, ATR can also directly phosphorylate p53 (6). P53 represses TCTP transcription through its binding to the p53 responsive element that is present in the TCTP promoter (7). TCTP directly interacts with MDM2 and stabilizes MDM2 (8), which results in increasing the MDM2-mediated ubiquitination of P53 and therefore p53 downregulation (9). P53 also promotes MDM2 expression by binding to its promoter (10)
Figure 3: A scheme of P53 � TCTP pathway during DNA repair
FIGURE 4
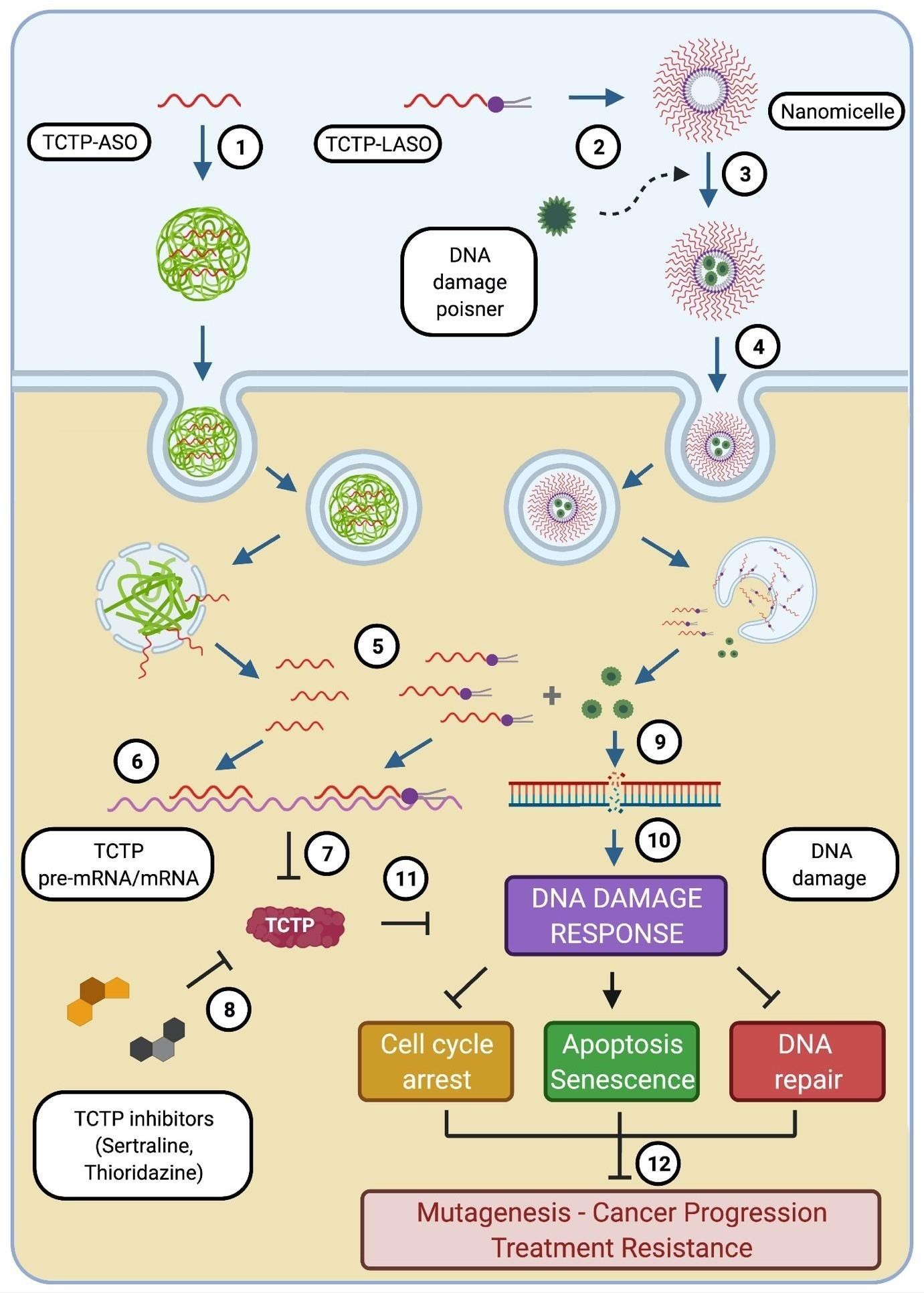
Antisense Oligonucleotide (ASO) targeting TCTP (called TCTP-ASO) is associated with a carrier (1), facilitating its cellular delivery. Alternatively, Lipid-modified TCTP ASO (TCTP-LASO) can be self- assembled into nanomicelle (2) which can encapsulate with a DNA damage poisoner (3) and go through the cell without delivery-aid (4). Inside the cell, ASO and LASO and encapsulated drugs can be released (5). ASO and LASO hybrid to the specific sequence of TCTP pre-mRNAs or mRNAs (6), leading to downregulation of TCTP protein (7). In addition, other small molecules (such as Sertraline and Thioridazine) also can be used to decrease TCTP protein level (8). These TCTP inhibitors (including TCTP-ASO and small molecules) can be used in parallel with DNA damage caused therapies (Irradiation, chemo drugs). On the other hand, the encapsulated DNA damage chemo-drugs released from the nanomicelles can induce DNA damages (9), which subsequently triggers DNA damage responses (DDRs) (10). TCTP downregulation interrupts DDRs (11), thereby blocking cell cycle arrest and DNA repair, promoting Apoptosis/Senescence, leading to decrease in possibilities of mutagenesis/cancer progression and treatment resistances (12)
Figure 4: Strategies of combinatorial treatment with TCTP inhibitors and DNA poisoners
Tables at a glance
Figures at a glance