PDGF Activates the DNA-PKcs-Dependent DNA Double-Strand Break Repair Pathway
Received Date: May 21, 2020 Accepted Date: June 10, 2020 Published Date: June 13, 2020
doi: 10.17303/jcvm.2020.6.202
Citation:Katrin Kramer (2020) PDGF Activates the DNA-PKcs-Dependent DNA Double-Strand Break Repair Pathway. J Cardio Vasc Med 8: 1-8.
Abstract
The atherosclerotic plaque milieu comprises growth factors and cytokines as well as DNA damage, which triggers inflammation, proliferation, and cell death. Previously, we had demonstrated that DNA-dependent protein kinase (DNAPK), an enzyme critically involved in DNA double-strand break (DSB) repair, is involved in human vascular smooth muscle cells (hVSMC) proliferation. In this study, we show that platelet-derived growth factor-BB (PDGF- BB), a predominant factor within the growing plaque, enhances DNA-PK catalytic subunit (DNA-PKcs) promoter activity with the subsequent transcriptional and translational upregulation of the protein. This upregulation of DNA-PKcs is important for the proliferation of hVSMCs. Additionally, PDGF-BB-dependent DNA-PK transactivation increased the capability of hVSMCs to repair DNA after treatment with ionizing radiation (IR). Through DNA-PK, PDGF-BB counterbalanced the deleterious effects of DNA DSBs on hVSMC integrity.
Keywords: DNA-PK; PDGF-BB; atherosclerosis; human vascular smooth muscle cells.
Abbreviations: NHEJ: Non-Homologous End Joining; DNA-PK: DNA-Dependent Protein Kinase; DNA-PKcs: The Catalytic Subunit of DNA-PK; hVSMC: Human Vascular Smooth Muscle Cell; MAP-kinase: Mitogen-Activated Protein Kinase; PI3K: Phosphatidylinositide 3-Kinases; PDGF-BB: Platelet-Derived Growth Factor Composed of Two B Subunits.
Introduction
DNA-dependent protein kinase (DNA-PK) is a nuclear serine/threonine kinase that belongs to the class IV phosphatidylinositol-3 (PI-3) kinase superfamily [1, 2]. It is composed of a DNA-PK catalytic subunit (DNA-PKcs) and a regulatory Ku70/ Ku80 heterodimer [3]. DNA- PK is a multifunctional enzyme. It is known as a DNA repair enzyme but it also accomplishes various other tasks [4, 5]. The special role of DNA-PK in response to IR is underlined by the fact that DNA-PK-deficient mice are more IR-sensitive. Here, the main function of DNA-PK is clear, namely, its important role in DSB repair [6].
There is increasing evidence that human atherosclerosis is associated with damage DNA [7]. Levels of DNA DSB have been shown to correlate with conventional cardiovascular risk factors in addition to being an independent predictor of the progression of atherosclerotic plaques [8]. Eukaryotic cells use two different ways to repair DNA lesions [9]. One of the mechanisms is homologous recombination, which is the repair of DSBs based on a homologous DNA strand [10]. Another way to repair DSBs and the major strategy in the context of DNA-PK is non-homologous end joining (NHEJ). NHEJ proceeds through three steps with DNA-PK as the central part: DSB recognition, processing of the DNA ends, and ligation of those ends. The Ku70/Ku80 heterodimer independently recognizes and binds the broken DNA ends by forming an asymmetric ring [11]. This complex then recruits the DNA- PKcs, which aligns the DNA ends. Because of a conformational change within the DNA- PKcs, it is autophosphorylated [12], which fulfills three important functions: the inactivation of DNA-PKcs kinase activity, the dissociation of the DNA-PK complex and thus the regulation and recruitment of enzymes in the processing step. During processing, the ends of the 5’ and 3’ DNA overhangs are modified so that subsequent ligation can occur. The involved enzymes complex with MRN [13] with the endonuclease Artemis and work in a complex with autophosphorylated DNA-PKcs [14, 15], phosphorylated XRCC4 [16], polynucleotide kinase phosphatase [17], tyrosyl-DNA phosphodiesterase 1 [18], the Werner syndrome helicase, which is activated through the phosphorylation of DNA-PK [19], and DNA polymerases µ and λ [20].
During our studies on the proliferation of hVSMCs, we were surprised to observe the upregulation of DNA-PKcs following treatment of the cells with the growth factor PDGF-BB. As hVSMCs are critical players in plaque growth, and their proliferation and survival potentially stabilize plaque, we hypothesized that PDGF-BB, in addition to its effect on proliferation, is able to modulate DNA-DSB formation in hVSMCs.
Materials and Methods
Cell culture: VSMCs from human aortic tissue explants containing atherosclerotic plaques were used and cultured at 37 °C in 5% CO2. After explantation, the endothelium and serosa of the vascular wall were removed, and the obtained muscular was digested with collagenase (CLS II, Biochrom, Berlin, Germany) and grown on culture plates. The experiments using human VSMCs were repeated with at least three independent cell preparations. In addition, we used a commercially available hVSMC line (#C-12533, PromoCell, Heidelberg, Germany). HVSMCs at passages 3–8 were used. The cells were exposed to irradiation from a BIOBEAM gamma irradiation device (Gamma-Service Medical, Leipzig, Germany). All procedures involving human materials had been approved by the local Ethics Committee and complied with the principles of the Declaration of Helsinki.
Transfection: Transfection was performed using a PromoFectin transfection kit (PromoCell, Heidelberg, Germany) according to the manufacturer’s instructions.
Antibodies: The following antibodies were used: antiDNA-PKcs (#sc-5282), (Santa Cruz Biotechnology, Heidelberg, Germany), anti-p-Akt-Ser473 (#4051), (New England Biolabs, Frankfurt/Main, Germany), anti-vinculin and anti-p-DNAPKcs-Ser2056 (#ab18192) (Abcam, Cambridge, UK), anti-p-γH2AX-Ser139 (#9718), anti-Chk2 (#3440) and Alexa Greenconjugated anti-mouse IgG (Molecular Probes, Eugene, OR, USA). A DNA Damage Sampler Kit (#9947) (New England Biolabs, Frankfurt am Main, Germany) was also used.
RNA Isolation and Real-Time RT PCR: After treatment, cell pellets were collected and total RNA was isolated with the Invisorb kit (Invitek, Berlin, Germany). cDNA synthesis was done with the Revert-Aid-H-cDNA synthesis kit (Fermentas, St. Leon-Rot, Germany). For real-time quantification, the SYBR Premix Ex Taq (Cambrex, Basel, Switzerland) was used. Quantitative real-time PCRs were performed using the following primers: DNA-PKcs forward, 5’ -GACTTGTACTCATCCTCACG-3, DNA-PKcs reverse, 5’ -GGGGCTTACCTGAGTGATCC-3’; CCND1, forward, 5’ -TTCATTGAACACTTCCTCTC-3’; CCND1 reverse: 5’-GTCACACTTGATCACTCTGG-3’; HPRT1 forward, 5’-TTGCGACCTTGACCATCTTTG -3’; HPRT1 reverse, 5’-CTTTGCTGACCTGCTGGATTAC-3’; Quantification was done using the ΔΔct-method.
Luciferase assay: Luciferase assays were performed using the luciferase assay system from Promega according to the manufacturer’s instructions and quantified with a luminometer (LB9506) (Berthold, Bad Wildbad, Germany).
Microscopy: Images of p-γ-H2AX-stained cells were acquired with an Axiovert 200M microscope (Carl Zeiss Microscopy GmbH, Jena, Germany) equipped with the fluorescence filter sets Zeiss No. 49 and HQ-FITC (EX BP 480/40, BS 505, EM 535/50) (Chroma Technology Corp., Bellows Falls, VT, USA), a scanning stage (Maerzhaeuser Wetzlar GmbH & Co. KG, Wetzlar, Germany) and an AxioCam MRm camera (Carl Zeiss Microscopy GmbH). Representative 1200 µm × 1200 µm areas per condition (6 × 8 fields of view or approximately 300 cells) were subsequently chosen, and the AxioVision module MosaiX, an EC Plan-Neofluar 40×/0.75 Ph2 objective lens, and constant imaging settings were used to acquire images to quantify the foci count. Subsequently, the images were exported in unaltered tagged image file format, and the image analysis program CellProfiler [21] was used to identify nuclei and γ-H2AX foci. Objects were related to each other, and the number of γ-H2AX foci per nucleus was measured and exported to a spreadsheet. For subsequent manual control, outlined overlay images were created.
Statistical Analysis
Data are given as mean ± SEM. Statistical analysis was performed by ANOVA. Post-test multiple comparisons was performed by the method of Bonferroni.
Results
The PDGF-induced proliferation of hVSMCs is DNA-PK-dependent
To analyze the influence of PDGF on hVSMCs, a proliferation assay was conducted (Figure 1A). Cells were treated with a specific inhibitor of DNA-PK, NU7026, and PDGF. The proliferation of hVSMCs increased during cell stimulation with PDGF compared to that of control cells. In contrast, cell proliferation decreased when the DNA-PK of hVSMCs was inhibited independently of PDGF stimulation (Figure 1A). We further tested the expression of cyclin D1 as a marker for proliferation and observed reduced protein expression following treatment of the cells with a DNA-PK inhibitor (Figure 1B). The protein expression results were confirmed by determining the mRNA expression of cyclin D1 (Figure 1C).
PDGF–dependent induction of DNA-PKcs
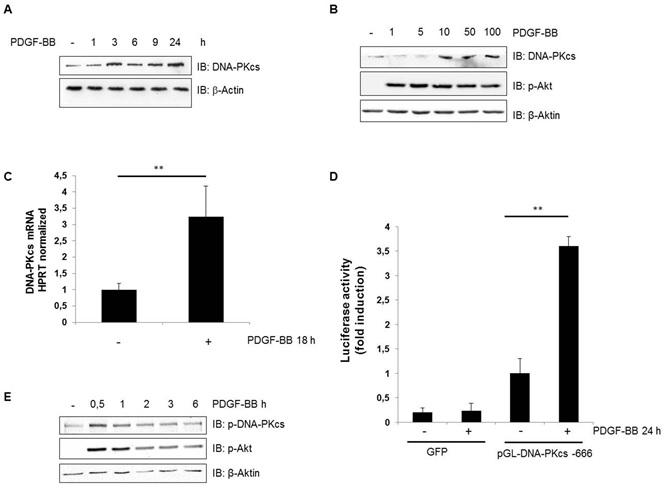
During our proliferation studies, we were surprised to see the upregulation of DNA-PKcs following the treatment of cells with PDGF-BB. Therefore, we studied the expression levels of DNA-PKcs after the treatment of hVSMC cells with PDGF-BB in a time-dependent manner and observed a marked upregulation of DNA-PKcs expression within 3 hours, which peaked 24 hours after the initiation of PDGF-BB treatment (Figure 2A). A dose-dependent upregulation of DNA-PKcs was observed, as well (Figure 2B). Furthermore, quantitative real-time PCR revealed that PDGF-BB induced the significant, approximately 4-fold induction of DNA-PKcs mRNA expression, which peaked 18 hours after treatment (Figure 2C). We were further interested in identifying transcription factors responsible for the upregulation of DNA- PKcs. Therefore, we cloned a fragment of the DNA-PKcs promoter (-3 to -606) [22] into the pGL3-Basic luciferase reporter plasmid and tested its promoter activity 24 hours after the transient transduction of human VSMCs. Transduction induced an approximately 3-fold increase in luciferase activity compared to that in control cells (Figure 2D).
In addition, using a phospho-specific antibody, we tested the phosphorylation of DNA-PKcs and observed increased phosphorylation within 0.5 hours of the treatment of hVSMCs with PDGF-BB (Figure 2E). This result suggested an increase in the activity of DNA-PKcs, as the phosphorylation of this serine is crucial for DNA-PKcs activity
PDGF-induced double-strand brake repair
The increased expression and activity of DNA-PKcs alone may provide important protection from genotoxic stress, even in cells already containing functional DNA-PK [23]. To determine the functional significance of our finding, we examined the effect of PDGF-BB on DSB repair in hVSMCs by studying the kinetics of γ-H2AX focus formation following the formation of DSBs during irradiation [24, 25]. Time-course experiments revealed that the number of γ-H2AX foci reached its maximum 1 hour after irradiation of hVSMCs (Figure 3A, B). Our result suggests that PDGF enhanced IR-induced DSB within 1 hour. Within 7 hours after irradiation, the number of γ-H2AX foci fell almost to the control level (Figure 3B). Foci formation was reduced significantly after 5 hours when cells had been pre-treated with PDGF- BB before irradiation (Figure 3B). The phosphorylation state of γ-H2AX was further studied in lysates of irradiated and PDGF-treated cells by immunoblot analyses. We found, significantly reduced phosphorylation of γ-H2AX after 5 hours compared to the phosphorylation of control cells without PDGF treatment; this confirmed the results from immunofluorescence microscopy (Figure 3A, B, C, D) Furthermore, the phosphorylation of Chk1 at Ser343 was used as a control to show successful irradiation, and the phosphorylation of Chk2 at Thr68 was used to show that DNA-PK had indeed been activated [26] (Figure 3E, F). In addition, we observed an increase of Chk1 phosphorylation after PDGF-BB treatment, indicating activation of the DNADSB repair pathway (Figure 3E).
Discussion
In addition to inflammation and proliferation, DNA lesions in cells promote atherosclerosis. These defects are caused not only by smoking or diabetes but also by reactive oxygen species [7, 27]. The role of DNA-PK in the pathogenesis of atherosclerosis was previously suggested [28]. Furthermore, we had demonstrated that DNA-PK is involved in VSMC proliferation [28]. The data of this study imply that the stimulation of VSMC proliferation by PDGF requires DNA-PK. In addition, our data demonstrate that DNA-PK mediates PDGF-BB- induced DSB repair. Initially, stimulation with PDGF-BB resulted in the increased appearance of γ-H2AX foci, which indicated the presence of DSBs. Subsequently, these DSBs were repaired more rapidly in the presence of PDGF-BB than in its absence. Our data suggest that PDGF induces DSBs and in turn activates DNA-PK to accelerate their repair. Furthermore, our data suggest two independent effects of the regulation of DNA-PK by PDGF-BB: first, rapid autophosphorylation of DNA-PKcs within 30 minutes of PDGF-BB treatment and second, an increase in DNA-PKcs within the following 3 hours. The rapid autophosphorylation of DNA-PKcs after PDGF-BB treatment suggests the increased activity of DNA-PK regarding its substrates. This upregulation of autophosphorylation can be explained by the results of a previous report [29], which demonstrated that the binding of transcription factors to DNA requires a break to unwind the DNA before proceeding, as does the autophosphorylation of DNA-PKcs. Therefore, considering this report, DNA-PK acts as a co-transcription factor in the regulation of PDGF-induced transcription. This is the first report to show that the treatment of cells with the growth factor PDGF-BB increases the level and activity of DNA-PKcs. Another growth factor, EGF, has also been reported to increase radio- resistance by activating DNA-PKcs [30, 31].
Elevated levels of DNA-PK indicate a fast and strong response to DNA damage, resulting in reduced cellular levels of DSBs. This rapid upregulation after PDGF-BB treatment indicates the direct binding of transcription factors to the DNA-PKcs promoter. A shortcoming of our study is that the last step of the PDGF signal transduction pathway, namely, the transcription factor responsible for DNA-PKcs upregulation, remains unknown. Using luciferase assays, we localized the DNA-PKcs promoter region (-666). However, further studies, for instance, the use of ChIP, are necessary to pinpoint this transcription factor.
These results shed new light on the regulation of DNAPK activity and imply that PDGF could counterbalance the deleterious effects of ionizing radiation on VSMC integrity.
Acknowledgements
We thank C. Zufelde for valuable technical assistance. Support from a Grant from the Deutsche Forschungsgemeinschaft (SFB854/A02) and GCI3, Medical Center, Otto-von-Guericke, is greatly appreciated.
- Kastan MB, and DS Lim (2000) "The many substrates and functions of ATM." Nature reviews. Molecular cell biology 1: 179-186.
- O'Driscoll M, et al. (2004) "An overview of three new disorders associated with genetic instability: LIG4 syndrome, RSSCID, and ATR-Seckel syndrome." DNA repair 3: 1227-1235.
- Lees-Miller SP, et al. (1990) "Human cells contain a DNAactivated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen." Molecular and cellular biology 10: 6472-6481.
- Komiyama S, et al. (2004) "Potentiality of DNA-dependent protein kinase to phosphorylate Ser46 of human p53." Biochemical and biophysical research communications 323: 816-822.
- Rinaldo C, et al. (2007) "MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damageinduced apoptosis." Molecular cell 25: 739-750.
- Downward J (2005) "RNA interference libraries prove their worth in hunt for tumor suppressor genes." Cell 121: 813-815.
- Mahmoudi M, et al. (2006) "DNA damage and repair in atherosclerosis." Cardiovascular Research 71: 259-268.
- Shah NR, and M Mahmoudi (2015) "The role of DNA damage and repair in atherosclerosis: A review." J Mol Cell Cardiol 86: 147-157
- Helleday T, et al. (2007) "DNA double-strand break repair: from mechanistic understanding to cancer treatment." DNA repair 6: 923-935.
- West SC (2003) "Molecular views of recombination proteins and their control." Nature reviews. Molecular cell biology 4: 435- 445.
- Walker JR, et al. (2001) "Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair." Nature 412: 607-614.
- Meek K, et al. (2008) "DNA-PK: the means to justify the ends?" Advances in immunology 99: 33-58.
- van den Bosch M, et al. (2003) "The MRN complex: coordinating and mediating the response to broken chromosomes," EMBO reports 4: 844-849.
- Ma Y, et al. (2002) "Hairpin opening and overhang processing by an Artemis/DNA- dependent protein kinase complex in nonhomologous end joining and V(D)J recombination." Cell 108: 781-794.
- Goodarzi A A, et al. (2006) "DNA-PK autophosphorylation facilitates Artemis endonuclease activity." The EMBO Journal 25: 3880-3889.
- Sharma MK, et al. (2016) "In cellular phosphorylation of XRCC4 Ser320 by DNA- PK induced by DNA damage." Journal of radiation research 57: 115-120.
- Weinfeld M, et al. (2011) "Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair." Trends in biochemical sciences 36: 262- 271.
- Kusumoto R, et al. (2008) "Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing." Biochemistry 47: 7548-7556.
- Ramsden DA (2011) "Polymerases in nonhomologous endjoining: building a bridge over broken chromosomes." Antioxidants & redox signaling 14: 2509-2519.
- Carpenter AE, et al. (2006) "CellProfiler: image analysis software for identifying and quantifying cell phenotypes." Genome Biology 7: R100.
- Connelly MA, et al. (1998) "The promoters for human DNAPKcs (PRKDC) and MCM4: divergently transcribed genes located at chromosome 8 band q11." Genomics 47: 71-83.
- Shen H, et al. (1998) "Increased expression of DNA-dependent protein kinase confers resistance to adriamycin." Biochimica et Biophysica Acta 1381: 131-138.
- Sedelnikova O A, et al. (2002) "Quantitative detection of (125) IdU-induced DNA double-strand breaks with the gammaH2AX antibody." Radiation research 158: 486-492.
- Rothkamm K and M. Löbrich (2003) "Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses." Proceedings of the National Academy of Sciences of the United States of America 100: 5057-5062.
- Li J and DF Stern (2005) "Regulation of CHK2 by DNA-dependent protein kinase." The Journal of biological chemistry 280: 12041-12050.
- Andreassi MG (2008) "DNA damage, vascular senescence and atherosclerosis." Journal of molecular medicine (Berlin, Germany) 86: 1033-1043.
- Medunjanin S, et al. (2015) "DNA-dependent protein kinase (DNA-PK) permits vascular smooth muscle cell proliferation through phosphorylation of the orphan nuclear receptor NOR1." Cardiovascular Research 106: 488-497
- Ju BG, et al. (2006) "A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription." Science (New York, N.Y.) 312: 1798-1802.
- Minjgee M, et al. (2011) "K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs." International journal of radiation oncology, biology, physics 81: 1506- 1514.
- Javvadi P, et al. (2012) "Threonine 2609 phosphorylation of the DNA-dependent protein kinase is a critical prerequisite for epidermal growth factor receptor-mediated radiation resistance." Molecular cancer research : MCR 10: 1359-1368.
Figures at a glance
 Figure 1
Figure 2
Figure 3
Figure 1
Figure 2
Figure 3
FIGURE 1
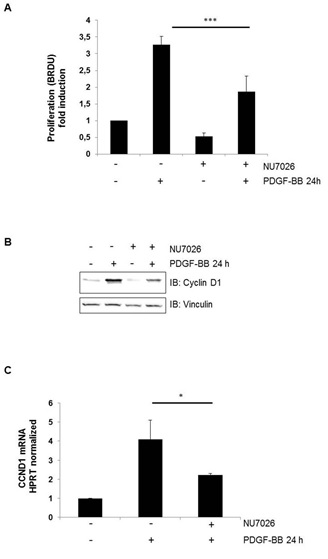
Figure 1: The PDGF-induced proliferation of hVSMCs is DNA-PK dependent. (A) Proliferation assay of hVSMCs treated with the DNA-PK inhibitor NU7026 (1 �mol/l) and PDGF-BB (10 ng/ml); BrdU incorporation was quantified. The fold induction is the ratio of stimulated to unstimulated cells. ***p < 0.001 (B) Immunoblot analysis of cyclin D1expression in hVSMCs after PDGF treatment and DNA-PK inhibition by the DNA-PK inhibitor NU7026; vinculin was used as a loading control. (C) mRNA expression of cyclin D1 in hVSMCs incubated for 6, 18, 24, and 48 hours with PDGF-BB (10 ng/ml) in the absence or presence of the DNA-PK inhibitor NU7026 (1 �mol/l). The fold induction is the ratio of stimulated to unstimulated cells, and error bars represent the S.D.s of three experiments (four measurements) *p < 0.05.
FIGURE 2
Figure 2: PDGF-dependent upregulation of DNA-PKcs. (A and B) Immunoblot analysis of DNA-PKcs expression in hVSMCs after PDGF-BB (10 ng/ml) treatment in a time- (A) and dose-dependent manner (B); �-actin was used as a loading control. (C) mRNA expression of DNA-PKcs in hVSMCs incubated for 18 hours with PDGF-BB (10 ng/ml); the fold change in induction is the ratio of stimulated to unstimulated cells. **p < 0.01. (D) Luciferase assay of hVSMCs after transfection of the pGL3-Basic luciferase reporter plasmid with and without a cloned fragment of the DNA-PKcs promoter (-3 to -606) treated with PDGF-BB (10 ng/ml). The fold induction is the ratio of stimulated to unstimulated cells, and error bars represent the S.D.s of three experiments (three measurements). **p < 0.01 (e). Immunoblot analysis of p- DNA-PKcs expression in hVSMCs after PDGF-BB treatment (10 ng/ml) in a time-dependent manner; �-actin was used as a loading control.
FIGURE 3
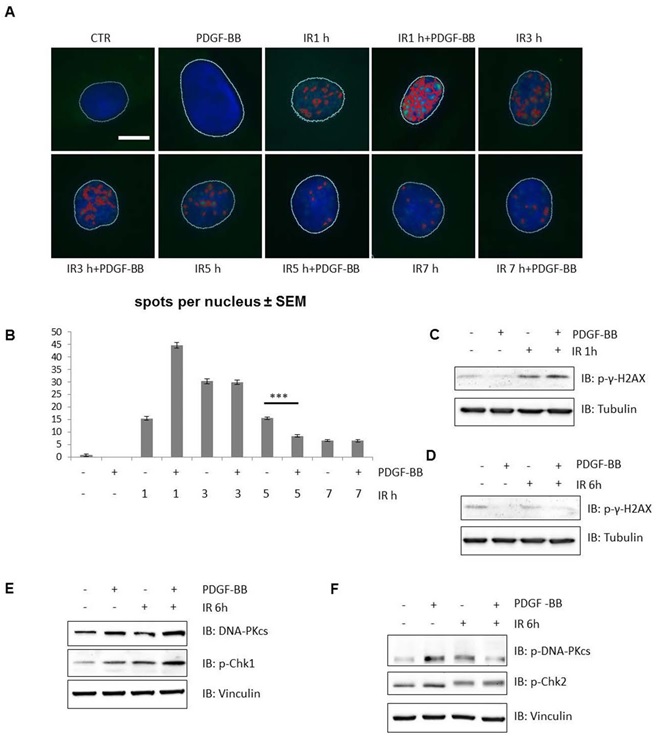
Figure 3: PDGF induces double-strand break repair. (A and B) (A) Immunofluorescence microscopy of ?-H2AX foci in nuclei and (B) a bar chart of ?-H2AX foci per nucleus as an indicator of double-strand breaks in hVSMCs treated with PDGF-BB and irradiated for 1, 3, 5 and 7 hours. ***p < 0.001. (C and D) Immunoblot analysis of phosphorylated ?-H2AX expression in hVSMCs incubated with PDGF-BB and treated with irradiation for 1 (C) and 6 (D) hours; tubulin used as a loading control. (E and F) Immunoblot analysis of DNA-PKcs (E) and phosphorylated DNA-PKcs (F) expression in hVSMCs treated with PDGF-BB and irradiated for 6 hours; phosphorylated Chk1 (E) and Chk2 (F) indicate DNA-PK activation; vinculin was used as a loading control.