A Comparative Study of Various SSRs Identification Tools Using Aspergillus Fu- migatus Chromosome Sequences
Received Date: July 26, 2020 Accepted Date: August 31, 2020 Published Date: September 02, 2020
doi: 10.17303/jbcg.2020.3.102
Citation:Manisha Mathur (2020) A Comparative Study of Various SSRs Identification Tools Using Aspergillus Fumigatus Chromosome Sequences. J Bioinfo Comp Genom 3:1-13.
Abstract
The objective of the present study is to compare different SSRs finding tool viz. MISA, MSATFINDER, SSR SERVER, SciRoKo, and TRF using the eight chromosomal sequences of a well-known fungus, Aspergillus fumigatus. Msatfinder and SSRServer provided with the results through email with all the desired information about the SSRs extracted, while the other three provided the results at the same time when the sequence is given to the tool as they are used offline. It was found that MISA, Msatfinder, SSRServer, SciRoKo are equally sensitive for all types of repeats but Tandem Repeat Finder (TRF) is highly sensitive for hexanucleotide repeat.
Keywords: Aspergillus fumigatus; SSRs Identification Tools; MISA; MSATFINDER; SSR SERVER; SciRoKo; TRF
Introduction
Simple sequence repeats (SSRs), also known as microsatellites, comprise tandemly repeated genetic loci of 1 to 6 base pairs(bp). SSRs are highly abundant and exhibit extensive levels of polymorphisms in eukaryotic [1, 2] and prokaryotic [3,4] genomes. SSRs are present in protein-coding and non-coding regions of the genome [2]. The use of SSRs or microsatellites as geneticmarkers has become very popular because of their abundance and length variation between different individuals. SSRs are highly polymorphic due to the high mutation rate affecting the number of repeat units and this hypervariability in SSRs has immense utility in different fields like genome characterization and mapping, preparation of linkage maps, phylogeny and evolutionary biology [5].
SSRs have several advantages over other molecular markers [6]. For example, (i) multiple SSR alleles may be detected at a single locus using a simple PCR based screen, (ii) they are evenly distributed all over the genome, (iii) they are co-dominant, (iv) a small quantity of DNA is required for screening, and (v) the analysis may be semi-automated and performed without the need of radioactivity. The potential biological function and evolutionary relevance of SSRs are currently under scrutiny and leading to a greater understanding of genomes and genomic initial suggestions that the majority of DNA was either 'junk' or had no biological function are being challenged by the discovery of new functions for these sequences. Several tools are freely available online to extract SSRs from the genome as well as EST sequences.
SRs were identified in various groups of organisms including plants [7,8], animals [9], fungi [10], etc. Aspergillus fumigatus is a well-known fungus, causes disease aspergillosis (a pulmonary infection), belongs to the genus Aspergillus of the phylum Ascomycota. Its entire genome of eight chromosomes has been sequenced and it has a genome size of ~30 Mb [11]. The abundance of SSRs (0.08% and 0.67% of the fungal genomes), provide a general source of molecularmarkers that could be useful for a variety of applications such as population genetics and strain identification of fungal organisms [11].
Apart from conventional methods [12, 13], several bioinformatics approaches exist for the identification of SSRs in the
available sequence data [14, 15, SSR server; Main lab Bioinformatics at Washington State University, 2003, 16]. MISA [14] allows the identification and localization
of perfect microsatellites as well as compound microsatellites,
which are interrupted by a certain number of bases. It also designs primers flanking the microsatellite loci, using two Perl
scripts. Large sequences are handled easily and input contains
two files: i)
Msatfinder [15] examines sequence and determines the number, type, and position of microsatellite repeats. There are two types of dependency required by Msatfinder: Perl modules, and external programs. Msatfinder is designed to find perfect repeats but in annotated (e.g. GenBank, EMBL, Swissprot) or unannotated (FASTA, raw) format files. It is also capable of finding interrupted microsatellites. It can be used to examine both protein and nucleic acid sequences. When run, Msatfinder creates some directories to store the output files. Results.html contains links to the contents of the following seven subdirectories: Repeats, GFF, Counts, Msat_tabs & Flank_tabs, Fasta, MINE, Primers. It can accept sequences up to 70Mb. Once the analysis is complete, a link to the results will be emailed to the user. Results are stored for 36 hours on the server before being deleted.
SSR server maintained by the Genome Database for Rosaceae (GDR), spans the sequence and filters out every possible SSR as per the parameters provided. It uses the modified SSRIT Perl script [13] with the FLIP program [17], which is a UNIX C program that translates and reformats DNA sequences in ORFs. Using the FLIP output, SSRIT selects the longest ORF as the putative coding region and reports the location of SSRs in the coding region. The results are provided to the user through the mail.
SciRoKo, SSR Classification and Investigation by Robert Kofler [18] is the combination of an extremely fast search algorithm with a built-in summary statistic tool which makes it an excellent tool for full genome analysis. This user-friendly software contains two modules: an SSR search module (supports five different SSR search modes) and an SSR-statistics module (for mismatch frequency and compound SSR). The SSR-search module is based on a scoring system that considers the length of a microsatellite. In the three perfect SSR search modes, a nucleotide at position i is tested for identity with the nucleotide at position 1+t, where t is the motif length (1–6). Upon identity, i is increased i+1 until no further identity can be found. If an identified SSR meets the specified minimum length (score), the SSR is saved to the output file [18].
Tandem Repeat Finder [16] is an algorithm for finding the tandem repeats which works without the need to specify either the pattern or pattern size. It models tandem repeats by percent identity and frequency of indels between adjacent pattern copies and uses statistically-based recognition criteria. In this, the user submits the sequence in FASTA format. The program is very fast, analyzing sequences on the order of 5Mb in just a few seconds. Submitted sequences may be of arbitrary length. Repeats with pattern size in the range from 1 to 2000 bases can be detected. The features supported by TRF are: (i) It uses k-tuple matching to avoid the need for full-scale alignment matrix computation, (ii) It requires no prior knowledge of pattern, pattern size or no. of copies, (iii) There is no restriction on the size of repeats, (iv) It determines a consensus pattern for the smallest repetitive unit in tandem repeats, and (v) There is an unlimited sequence size. After program execution, two files are returned, the first is a summary table describing the location and statistical properties of tandem repeats found and the second contains the alignment of each repeat with its consensus sequence.
Considering the points discussed above the objective of the present study is to compare different SSRs finding tool viz. MISA, MSATFINDER, SSR SERVER, SciRoKo, and TRF using the eight chromosomal sequences of a well-known fungus, Aspergillus fumigatus.
Material and Methods
1. Retrieval of Chromosome Sequences of Aspergillus fumigatus
Sequences of eight completely sequenced chromosomes of Aspergillus fumigatus (NC_007194, NC_007195, NC_007196, NC_007197, NC_007198, NC_007199, NC_007200, and NC_007201) were retrieved from National Center for Biotechnology Information (NCBI; ftp://ftp.ncbi.nih.gov/genomes/Fungi/Aspergillus_fumigatus/).
2. Harvesting of Simple Sequence Repeats Using Different Tools
The SSRs were extracted out of the sequences using the several available tools for finding SSRs in the genome sequences. Many freely available tools were studied and then finally the five tools were selected to accomplish the task, based upon their capacity, platform supported and suitable working algorithm to compare the data obtained in the form of SSRs.
The selected tools are SSR SERVER, TANDEM REPEAT FINDER, MISA, MSATFINDER, and SciRoKo. The parameters of different tools taken in the present study are as follows:
Results
In the present study 8 chromosomal sequences of Aspergillus fumigatus available at NCBI (https://www.ncbi.nlm.nih. gov) were screened for simple sequence repeats. SSRs were detected in 8 chromosomes using MISA, Msatfinder, SSRServer, SciRoKo, and Tandem Repeat Finder. Msatfinder and SSRServer provided with the results through email with all the desired information about the SSRs extracted, while the other three provided the results at the same time when the sequence is given to the tool as they are used offline. The tools extracted the SSRs out of the genome sequence of Aspergillus fumigatus, in the form of repeated motifs and their corresponding length in the sequences. The SSRs contained in each chromosomal sequence, obtained from different tools were compared with each other and looked for their presence or absence in tools considered. All poly-A and poly T repeats were not considered as SSRS due to their presence at 3'end of mRNA/cDNA sequences.
Compiled results represent the frequencies of all types of repeats in the whole genome obtained from selected tools is shown in the summary table.
Discussion
In the recent past, a number of studies on the discovery and use of SSRs in several species [20, 19, 9, 22, 7, 17] using traditional [15] and bioinformatics mining approaches [16] have been undertaken. The present study examining and comparing SSRs finding tools to check their efficiency on the basis of different types of repeats by using a completely sequenced Aspergillus fumigatus genome.
In the present study, the genome of A. fumigatus in the form of chromosomal sequences (8 chromosomes) has been retrieved from Genome Assembly/Annotation Projects Database maintained at NCBI and available at https://www.ncbi.nlm.nih. gov. From these chromosomal sequences, SSRs were mined and characterized using freely available tools MISA, Msatfinder, SSRServer, SciRoKo, and Tandem Repeat Finder (TRF) for extracting the SSRs from the genome sequence. The tools are selected on the basis of the following criteria, i) Which can mine SSRs in chromosomal sequences of moderate size ii) Find perfect repeats with their location in the sequence. iii) Satisfy our working environment and iv) Mine SSRs timely.
The data obtained from all the tools were compared for the repeat type and their frequencies obtained, so as to check the efficiency and suitability of the tools while harvesting of SSRs, keeping minimum repeat length 14 and motifs 1-6 bp long, i.e. all the programs were compiled using the same level of optimization.
Different tools reporting different frequency of repeats for different repeat types as MISA, Msatfinder, SSRServer, SciRoKo, and TRF have given 1622, 1617, 1621, 1606, and 271 respectively. Thus, further studies can be done to resolve the significance of the variation in the number of repeat types of SSRs in the genome of A. fumigatus and thus can also be applied to the annotation of the genome of other organisms too, as SSRs act as the markers in the genome.
Mononucleotide repeats are found in the A. Fumigatus genome with relatively low frequency as a comparison to other repeat types. Poly A & poly T repeats have been excluded due to which their number is underrepresented in the study [17]. Among all the tools, TRF has given the least monorepeats while rest have given approximately the same results. 3.03 repeats/ Mb, 3.0 repeats/Mb, 3.03 repeats/Mb, 3.03 repeats/Mb and 0.1 repeats/Mb mononucleotide repeats have been extracted by MISA, Msatfinder, SSR server, SciRoKo and TRF respectively. However, the frequency of mononucleotides detected in this study was lower than the frequencies of SSRs found in fungal genomes: 3,063 repeat/Mb in M. grisea, 2,505 repeats/Mb in N. crassa,2,071 repeats/Mb in S. cerevisiae and 156 repeats/Mb in E. cuniculi [10].
It has been reported that AT and CT are the most common repeat motif in plants and insects [13]. In our study AT repeats are predominant in the genome of A. fumigatus. The higher AT frequenciesin the genome can be assumed to be the result of the high A/T content of the genomes [10]. 7.21 repeats/Mb, 7.21 repeats/Mb, 7.21 repeats/Mb, 7.14 repeats/Mb, and 1.25 repeats/ Mb dinucleotide repeats have been extracted by MISA, Msatfinder, SSR server, SciRoKo, and TRF respectively. The densities of dinucleotides between the genomes varied significantly,with 552.5 repeat/Mb in N. crassa having the highest and 141 repeat/ Mb in A. nidulans the lowest densities [10].
Trinucleotide repeats are highly abundant in the A. fumigatus genome according to the results given by MISA, Msatfinder, SSRServer and Sciroko. This abundance of tri-nucleotide repeats is in agreement with the results of earlier studies in N. crassa,U. maydis [10]. 16.29 repeats/Mb, 16.29 repeats/Mb, 16.29 repeats/Mb, 16.15 repeats/Mb and 2.61 repeats/Mb trinucleotide repeats have been extracted by MISA, Msatfinder, SSR server, SciRoKo, and TRF respectively.
The density of tetranucleotide repeats was found to be much lower than the densities of dinucleotide and trinucleotide repeat motifs in all tools used during the study. The lowest relative abundance of tetranucleotide repeats was also reported in S. cerevisiae (one repeat/Mb), and E. cuniculi. The most frequent tetranucleotide repeat motifs were much less in abundance than the lower-repeated units [10]. Generally, most tetranucleotide repeats were rather short, with a repeat length of less thaneight.4.08 repeats/Mb, 3.97 repeats/Mb, 4.08 repeats/Mb, 3.97 repeats/Mb and 0.91 repeats/Mb tetranucleotide repeats have been extracted by MISA, Msatfinder, SSR server, SciRoKo, and TRF respectively.
Theoretically, the occurrence of pentanucleotide repeats should be less than that of the tetranucleotide repeats as in N. crassa [10] but in A. fumigatus the frequency of pentanucleotides 10.23 repeats/Mb, 10.27 repeats/Mb, 10.20 repeats/Mb, 10.13 repeats/Mb and 0.91 repeats/Mb given by MISA, Msatfinder, SSR server, SciRoKo, and TRF respectively, is not following the above concept.
In A. fumigatus, hexanucleotides were the second abundant repeat type followed by Penta, di, tetra and mononucleotide repeats Moreover, in the genomes of C. neoformans, M. grisea, S. cerevisiae, and U. maydis hexanucleotide repeats were more abundant than the pentanucleotides [10]. MISA, Msatfinder, SSR server, SciRoKo and TRF have been detected 14.31 repeats/Mb, 14.25 repeats/Mb, 14.31 repeats/Mb, 14.21 repeats/Mb and 3.6 repeats/Mb hexanucleotide respectively. TRF screened a higher percentage of hexanucleotide in comparison to other repeat types.
However, the frequency of SSRs detected in the present study was lower than the frequencies of SSRs detected in nine fingal species, [10] in all other fungal genomes that belong to the same phylum Ascomycota. This variation occurs due to Among all the tools TRF reported the lowest number of repeats due to lack of desired flexibility in the parameters and it is the only tool whose algorithm allows indels (insertion and deletion) due to which imperfect SSRs are reported which are not needed.
Conclusion
A qualitative study of various simple sequence repeats identification tools for quantitative detection of SSRs in the whole genome sequences has been successfully conducted. A set of freely available tools (MISA, Msatfinder, SSRServer, SciRoKo, Tandem Repeat Finder (TRF)) has been tested for their sensitivity to detect different types of SSRs. It was found that MISA, Msatfinder, SSRServer, SciRoKo are equally sensitive for all types of repeats but Tandem Repeat Finder (TRF) is highly sensitive for hexanucleotide repeat.
- Tautz D, Renz M (1984) Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 12: 4127-4138.
- Katti MV, Ranjekar PK, Gupta VS (2001) Differential distribution of simple sequence repeats in the eukaryotic genome sequence. Mol. Bio. Evol. 18: 1161-1167.
- Field D, C Wills (1996) Long, polymorphic microsatellites in simple organisms. Proc. R. Soc. Lond. B Biol. Sci. 263: 209–215.
- Gur-Arie R, CJ Cohen, Y Eitan, L Shelef, EM Hallerman, Y Kashi (2000) Simple sequence repeats in Escherichia coli: abundance, distribution, composition, and polymorphism. Genome Res. 10: 62–71.
- Grover A, Aishwarya V, Sharma PC (2007) EuMicroSatdb: A database for microsatellites in the sequenced genomes of eukaryotes. BMC Genomics 8: 225.
- Edwards D, Robinson AJ, Love CG, Batley J (2003) Simple sequence repeat marker loci discovery using SSR primer. Bioinformatics 20: 1475–1476.
- Kantety R, Rota M, Mathews D, Sorrells M (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum, wheat. Plant Mol Biol. 48: 501-510.
- Shanker A, Singh A, Sharma V (2006) In silico mining in the expressed sequence of Neurospora crassa for identification and abundance of microsatellites. ScienceDirect 162: 250-256
- Serapion J, Kucuktas H, Feng J, Liu Z (2004) Bioinformatics Mining of Type I Microsatellite from expressed Sequence Tags. Mar. Biotechnol 6: 364-377.
- Karaoglu H, Lee CMY, Meyer W (2004) A Survey of simple sequence repeats in completed fungal genomes. Mol. Bio. Evol. 22: 39-49.
- Nierman WC, Pain A, Anderson MJ, Wortman JR, et al. (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438: 22-29.
- Beckmann JS, Soller MTowards (1990) A unified approach to the genetic mapping of eukaryotes based on sequencetagged microsatellite sites. Bio/Technology 8: 930-932.
- Temnykh S, DeClerck G, Lukashova A, Lipovich L, et al. (2001) Computational and experimental analysis of microsatellites in rice: frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 11: 1441-1452.
- Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 106: 411-422.
- Thurston MI (2005) Field D: Msatfinder: detection and characterization of microsatellites.
- Benson G (1999) Tandem Repeat Finder: A program to analyze DNA sequences. Nucleic Acids Res. 27: 573-580.
- Bossard N (1997) FLIP: a Unix program used to find/ translate ORFs. Bionet Software.
- Kofler R, Schlotterer C, Lelley T (2007) SciRoKo: A new tool for whole genome microsatellite search and investigation.
- Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol. Biol. Lett. 7: 537-546.
- Gupta PK, Rustagi S, Sharma S, Singh R, Kumar N, Balyan HS (2003) EST-SSRs for transferability, polymorphism, and genetic diversity. Mol. Genet. Genomics 270: 315-323.
- Sekinoe M, Hamaguchi M, Aranishi F, Okoshi K (2003) Development of novel microsatellite DNA markers. Mar Biotechnol 5: 227-233.
- Lagercrantz U, Ellegren H, Andersson L (1993) The abundance of various polymorphic microsatellite motif differs between plants and vertebrates. Nucleic Acids Res. 21: 1111- 1115.
- Website References https://tandem.bu.edu/trf/trf400.win.download.html https://bioweb.pasteur.fr/seqanal/interfaces/equicktandem.html https://bioweb.pasteur.fr/seqanal/interfaces/etandem.html https://www.gramene.org/db/searches/ssrtool https://pgrc.ipk-gatersleben.de/misa/ https://espressosoftware.com/pages/sputnik.jsp https://bibiserv.techfak.uni-bielefeld.de/reputer/ https://www.mainlab.clemson.edu/cgi-bin/gdr/gdr_ssr https://bioinformatics.org/poly/wiki/ https://bioweb.pasteur.fr/seqanal/interfaces/mreps.html https://atgc.lirmm.fr/star/ https://www.drive5.com/piler https://www.genomics.ceh.ac.uk/msatfinder/ https://www.Kofler.or.at/Bioinformatics https://www.cdfd.org.in/imex https://www.biophp.org/minitools/microsatellite_repeats_finder/ demo.php https://hornbill.cspp.latrobe.edu.au/ssrdiscovery.html https://www.irri.org/science/abstracts/004.asp
 Figure 1
Figure 2
Figure 3
Figure 4
Figure 5
Figure 6
Figure 7
Figure 8
Figure 9
Figure 10
Figure 1
Figure 2
Figure 3
Figure 4
Figure 5
Figure 6
Figure 7
Figure 8
Figure 9
Figure 10
FIGURE 1
Figure 1: Frequency Variation in Repeat Types of Genome of A. fumigatus using different Tools.
FIGURE 2
Figure 2: Frequency Variation in Repeat Types of chromosome 1 of A. fumigatus using different Tools.
FIGURE 3
Figure 3:Frequency Variation in Repeat Types of chromosome 2 of A. fumigatus using different Tools.
FIGURE 4
Figure 4: Frequency Variation in Repeat Types of chromosome 3 of A. fumigatus using different Tools.
FIGURE 5
Figure 5: Frequency Variation in Repeat Types of chromosome 4 of A. fumigatus using different Tools.
FIGURE 6
Figure 6: Frequency Variation in Repeat Types of chromosome 5 of A. fumigatus using different Tools.
FIGURE 7
Figure 7:Frequency Variation in Repeat Types of chromosome 6 of A. fumigatus using different Tools.
FIGURE 8
Figure 8: Frequency Variation in Repeat Types of chromosome 7 of A. fumigatus using different Tools.
FIGURE 9
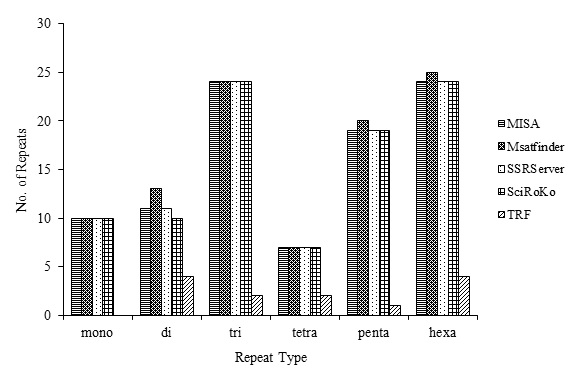
Figure 9:Frequency Variation in Repeat Types of chromosome 8 of A. fumigatus using different Tools.
FIGURE 10
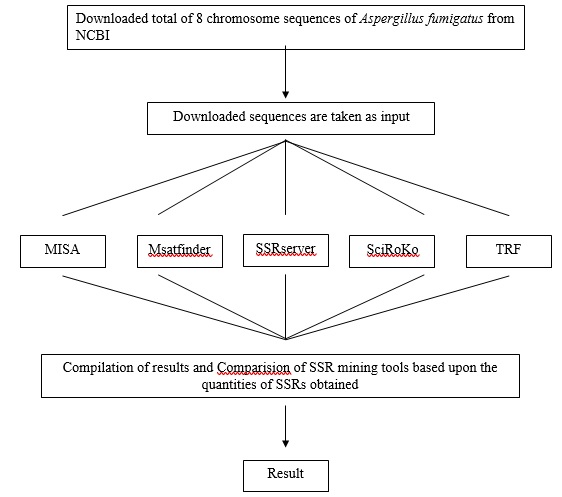
Figure 10:Flowchart represents a methodology of work
Tables at a glance