Activation of P53 Via Nutlin-3a Reveals Role for P53 In ROS Signaling During Cardiac Differentiation of hiPSCs
Received Date: February 25, 2021 Accepted Date: March 25, 2021 Published Date: March 27, 2021
doi: 10.17303/jscr.2021.3.101
Citation: Emma B Brandt (2021) Activation of P53 Via Nutlin-3a Reveals Role for P53 In ROS Signaling During Cardiac Differentiation of hiPSCs. J Stem Cell Rep. 3: 1-22.
Abstract
Activation of the transcription factor P53 within cancer cells is a well-characterized pathway, whereas the effects of P53 activation during development remain largely unexplored. Previous research has indicated that increased levels of P53 protein during key murine developmental stages cause defects in multiple embryonic tissues, including the heart. These findings were confirmed in several different mouse models of congenital heart defects, but P53 activation in a human system of cardiovascular development is not available. Utilizing human induced pluripotent stem cells (hiPSCs), we characterized the normal levels of P53 during cardiac differentiation and showed that levels of P53 are high in hiPSCs and decrease upon cardiac lineage commitment. We also observed P53 localization changed from mainly cytoplasmic in iPS colonies to the nucleus in the Nkx2-5 + cardiac progenitor stage. Pharmacological-mediated increase of P53 protein levels with the Mdm2 inhibitor Nutlin-3a during early (mesoderm to cardiac mesoderm) stages of cardiogenesis resulted in a sizeable loss of cardiomyocytes due to increased apoptosis and cell cycle arrest. Interestingly, increasing P53 levels did not result in apoptosis at later (cardiac progenitor to beating cardiomyocytes) stages of the cardiac differentiation. These results illustrate the temporal sensitivity to increased P53 levels during cardiogenesis. We conducted RNA-Seq on these cells with or without Nutlin-3a to ascertain transcriptional differences due to increased P53 at the different stages during the differentiation. Our results from the RNA-Seq revealed up-regulation of Sestrins after Nutlin-3a treatment suggesting a new role for P53 in the metabolism of cardiac regeneration.
Keywords: Human induced pluripotent stem cells; P53; Cardiac differentiation; Reactive oxygen species
Introduction
Most research regarding P53 has focused on its loss and subsequent role in tumorigenesis. From a developmental perspective, loss of P53 revealed few phenotypes but classically resulted in increased cancer susceptibility [1,2]. Interestingly, the loss of P53’s main regulator Mdm2 was embryonic lethal and was rescued by concomitant deletion of a P53 allele [3,4]. The role of Mdm2 as a ubiquitin ligase for P53 degradation revealed a critical need for P53 regulation during development in multiple tissue types [5,6] including cardiac. Further work demonstrated that elevated P53 levels due to cardiac-specific Mdm2 +/- or Mdm2 -/fm alleles caused increases in cell death and decreases in cell proliferation during murine cardiogenesis, ultimately leading to cardiac malformations such as atrial and ventricular septal defects [7,8]. These studies revealed that regulation of P53 by Mdm2 during development is crucial for embryonic survival.
Additionally, loss of a P53 allele was beneficial in alleviating phenotypes of several congenital heart defect (CHD) models such as DiGeorge or CHARGE syndromes [9-11]. Early stages of cardiogenesis are characterized by distinct processes of cell migration and differentiation critical for proper looping and chamber formation. Loss of cells during these initial stages of cardiogenesis would have detrimental outcomes and could lead to cardiac malformations.
Stress signaling pathways in cardiac tissues in response to environmental influences should also be considered when evaluating CHD pathogenesis. Exogenous stressors applied to the developing heart such as X-ray radiation 8 or high glucose [12] resulted in increased P53 levels that contributed to CHD development.
Thus, although increased P53 activity is clearly associated with CHD pathogenesis, P53 signaling in early stages of cardiac development remains largely unexplored.
Human induced pluripotent stem cells (hiPSCs) present a powerful model system to help model early human development in a dish and generate specific sub-types of human cells typically unavailable for research [13,14] Cardiac differentiation of hiPSCs allows for observation of cells from early human cardiac formation and these can help provide insight into how some human congenital heart defects arise. Additionally, understanding how in vitro cardiac differentiation stimulates cell stress responses will be useful in the development of future generations of healthy cardiomyocytes, along with understanding the limitations of this system.
There is currently little research regarding P53’s role in cardiac differentiation. The activity of P53 regarding maintenance of pluripotent stem cells (PSCs) has been well established in cell survival and differentiation abilities. Notable, that PSCs are unusually sensitive to increased levels of P53, and in response to small molecules such as Nutlin-3a or irradiation, they undergo rapid apoptosis or differentiation from the pluripotent state [15-20].
Other work showed loss of P53 resulted in difficulties differentiating and affected the cells’ ability to balance repression of pluripotency genes and activation of differentiation-related genes [21]. The P53 family of transcription factors (P53, P63, and P73) were determined to be critical regulators of Wnt signaling in mesendoderm formation through interactions with Smad nuclear co-regulators [22]. Another study found that genetic deletion of P53 inhibited cardiogenesis in mouse ESCS. This loss of P53 resulted in prolonged Nanog expression in the differentiated cells leading to reduced levels of mesoderm and cardiac genes ultimately causing reduced cardiac yields [23]. In summary, these studies illustrate that loss of P53 impaired the ability of pluripotent stem cells to differentiate into cardiac lineages.
Conversely, increased P53 expression in patient-derived cardiac mesenchymal stromal cells (similar to dermal mesenchymal stromal cells regarding expression of CD73, CD90, and CD105) was shown to increase their cardiac differentiation capacity after HDAC1 depletion [24]. Thus, the timing of P53 activity seems to be critical for cardiogenesis, but modulation of P53 levels during the distinct stages of in vitro cardiac differentiation has yet to be studied. We hypothesized that activation of P53 would produce disparate results on cell survival and proliferation at distinct developmental-based cell types in our cardiac differentiation protocol.
To investigate the effects of active P53 in early cardiogenesis, we utilized human induced pluripotent stem cells (hiPSCs) for cardiac differentiation and treated the cells at progressive stages of differentiation with an Mdm2 specific antagonist Nutlin-3a [25]. Application of the small molecule inhibitor Nutlin-3a allowed us to rapidly increase P53 protein levels and target specific cardiac developmental lineages during cardiac differentiation. The resulting experiments revealed early stages of cardiac differentiation (iPSCs and mesoderm) had increased apoptosis when P53 levels were elevated, but cardiac progenitors and cardiomyocytes lost this apoptotic response. We conducted RNA-Seq on mesoderm, cardiac progenitors, and cardiomyocytes with or without exposure to Nutlin-3a and found a non-canonical role for P53 in cellular oxidant handling during cardiac differentiation.
Materials and Methods
Human induced pluripotent stem cell (hiPSC) derivation and culture
hiPSCs were generated by ReGen Theranostics (Rochester, MN) using patient primary fibroblasts from healthy individuals who were de-identified according to institutional guidelines (Mayo Clinic IRB 10-006845, Clinical Trials Identifier NCT01860898). The fibroblasts were reprogrammed using CytoTune-iPS Sendai Reprogramming Kits (Invitrogen) following the manufacturer’s instructions. Any residual fibroblast culturing used DMEM + Glutamax (Gibco)/ + 10% FBS. hiPSCs were cultured using mTeSR (Stem Cell Technologies) and plated on Geltrex (Thermo Fisher). Colonies were passaged and maintained using ReLeSR (Stem Cell Technologies).
Pluripotency was assessed via qRT-PCR for pluripotency markers (Oct4, Sox2, Nanog, and c- Myc) as compared to their original fibroblasts. Immunofluorescence for pluripotency markers was conducted on hiPSCs plated with Geltrex (Thermo Fisher) on Permanox chamber slides (Thermo Fisher), stained with Pluripotency Stem Cell 4-Marker ICC Kit (Invitrogen) according to the manufacturer’s instructions, and imaged on a LSM 780 inverted confocal microscope (Carl Zeiss). Etoposide sensitivity assays [26] were conducted on the original fibroblasts, along with each iPSC clone used for an individual as reported previously with a sensitivity cut off of 100 nM. Mitochondrial DNA (mtDNA) sequencing was also performed to eliminate any mtDNA mutations in the clones used that could have arisen during the reprogramming process [27].
in vitro cardiac differentiation
For cardiac differentiation, hiPSCs were enzymatically split using TrypLE (Thermo Fisher) and resuspended in mTeSR plus 10 nM ROCK inhibitor (R&D Systems) and subsequently re-plated as single cells in a monolayer prior to initiating cardiac differentiation. We used the chemical defined media (CDM3) cardiac differentiation protocol as previously published [28]. Briefly, cells were initiated with 8 μM CHIR (Selleck Chem) and 10 nM Activin A (R&D Systems) for 20 hours. Medium was then removed and replaced with CDM3 for an additional 24 hours. On Day 3 cells were treated with 5 μM IWP2 (R&D Systems) and 10 nM BMP4 (R&D Systems) for another 48 hours. Cells were maintained in CDM3 with medium changes every 2 days. Spontaneous beating was typically observed around Day 8. Beating cardiomyocytes were harvested using Liberase TH Research Grade (Creative Enzymes/Roche) and DNAse (Roche) for digestion of ECM, then made into a single cell suspension using TrypLE (Thermo Fisher) for either re-plating or flow cytometry. If cardiomyocytes were re-plated, plates were first coated with Fibronectin (Sigma Aldrich).
Drug treatments
All drugs were dissolved in DMSO at the following concentrations: Nutlin-3a (Selleck Chem) working stocks were stored at 50 mM or 10 mM, Z-VAD-FMK (Selleck Chem) was stored at a 50 mM, and SJ172550 (Santa Cruz) was stored at a 10 mM stock concentration. All drugs were stored at -20 °C, DMSO was employed as a vehicle control, and the final DMSO concentration was maintained at or below 0.1%. Dose curves for each drug were performed on Day 5 of cardiac differentiation to determine the appropriate working concentration.
qT-PCR
Total RNA was isolated from hiPSCs using Trizol (Life Technologies) and RNeasy mini kits (Qiagen). cDNA was synthesized from 1 μg of RNA using iScript cDNA Synthesis Kit (Bio-Rad). Taqman Universal PCR Master Mix (Applied Biosystems) and 10-15 ng of cDNA underwent 40 cycles of PCR using a ViiA7 Real-Time PCR System (Thermo Fisher) with Taq- Man (FAM-labeled) probes and primers (IDT). GAPDH and/or β-actin were included as internal loading controls. (See Supplementary Table 1)
Western blotting
Cells were washed with ice-cold PBS, scraped into suspension, then lysed in RIPA buffer for 20-30 minutes. Protein lysate was quantified using a Pierce BCA Assay (Thermo Fisher), then separated via SDS-PAGE in Criterion gels (Bio-Rad). Total protein was stained for using REVERT total protein stain kit (LI-COR) immediately following transfer.
Membranes were blocked in TBS Blocking Buffer (LI-COR) and then incubated with the TBS Blocking buffer plus 0.2% Tween with the following primary antibodies: P53 (1:1000, Abcam ab1101), Mdm2 (1:500, Abcam ab38618), P53-mediated apoptosis antibody cocktail (1:300, Abcam ab140360), Noxa (1:1000, Enzo Life Sciences ALX-804-408-C100), Puma (1:300, Santa Cruz sc-374223), Mcl1 (1:1000, Cell Signaling 4572S), Bcl-xL (1:1000, Cell Signaling 2764S), Bcl2 (1:100, Thermo Fisher MA1-26233), and Vinculin as a loading control (1:1000, Abcam ab73412 & ab129002). Blots were rinsed with TBST 4 times for 5 minutes.
Finally, the membrane was probed with fluorescent secondary antibodies (IRDye 800CW and IRDye 680RD, 1:15,000) (LI-COR) in TBS Blocking Buffer (LI-COR) + 0.2% Tween + 0.01% SDS and imaged on an Odyssey Clx Imaging System (LI-COR).
Immunofluorescence
Cells were plated on Permanox plastic chamber slides (Thermo Fisher) coated with Geltrex (Thermo Fisher) or Fibronectin (Sigma Aldrich). The cells were fixed with 4% PFA-2% glucose and permeabilized in a solution composed of 0.5% Triton X-100 (Sigma T8787)-20 mM HEPES(pH 7.4)/50 mM NaCl/3 mM MgCl2/300 mM sucrose. Slides were blocked with Superblock (Thermo Fisher) for 3 hours at RT. Primary antibodies were incubated in PBS/10% Superblock (Thermo Fisher)-0.1% Tween for several hours at RT or overnight at 4 °C: P53 (Abcam ab1101 or ab179477), Tra-1-60 (Thermo Fisher 41-1000), Nanog (Cell Signaling 3580), NKX2-5 (Cell Signaling 8972S), α-actinin (Abcam ab137346). The slides were then washed with PBS + 0.1% Tween three times then incubated with the appropriate secondary antibodies for 1 hr at RT in the dark. The slides were washed twice more with PBS + 0.1% Tween and then twice with PBS alone. Nuclei were stained with DAPI, specimens mounted with Prolong Gold Antifade (Fisher Scientific) and covered with a 25 mm coverslip. The slides were then imaged on a LSM 780 inverted confocal microscope (Carl Zeiss) at 10-, 20-, and 40X at the Mayo Microscopy and Cell Analysis Core.
Flow cytometry
For the EdU analysis, cells were incubated with 10 μM EdU supplied with the Click-iT Alex Fluor 555 Imaging Kit (Thermo Fisher) for 1 hr. Cells were harvested with TrypLE or Liberase/DNAse treatment and made into a single cell suspension. EdU stained single-cells were incubated with a Live-Dead viability dye (NearIR 755 nM or Brilliant Violet 395 nM excitation λ, Thermo Fisher) at 37 °C for 20 minutes. Cells were then fixed with 4% PFA, resuspended in FACS( DPBS 1X, 0.5% fetal bovine serum, 2mM EDTA, pH 7.4) buffer, and stored at 4°C in the dark. Prior to running the cytometer, cells were washed and permeabilized (BD Cytofix/Perm kit). Cells incubated with EdU were stained with Alexa Fluor 555 conjugated Click-iT molecular probes (Thermo Fisher). Antibodies towards intracellular antigens cardiac markers cTnI (BD Biosciences, 564409) and CtnT (BD-Biosciences, 564767) were stained at the same time as the EdU. Nuclei sometimes were stained with DAPI and then the cells were run on a Beckman Coulter Flow Cytometer and data was analyzed using Kaluza.
RNA-Seq
The hiPSC derived cardiac differentiation was performed as described above, and RNA was isolated from one control cell line with 8 biological replicates for Day 3, Day 5, Day 7, and Day 10 (6 replicates) treated with DMSO or 10 uM Nutlin-3a for 24 hours. Total RNA was isolated using Trizol (Life Technologies) and RNeasy mini kits (Qiagen). RNA samples were submitted to the Mayo Genome Facility Core for RNA sequencing where they were assessed a RIN (RNA integrity number) score, and then sequenced on an Illumina sequencing platform using paired-end index reads. Raw RNA-Seq data (FASTQ files) were analyzed using Mayo Bioinformatics Core standard RNA-seq pipeline for mapping the reads to the human transcriptome to quantify gene expression and generate the RPKM matrix [29]. To take advantage of this time-course gene expression data, the RPKM values of all genes were analyzed using a generalized linear model (GLM) to identify genes differentially expressed between the control and treated time course data. Gene fold changes were calculated pairwise at each time point. The differentially expressed genes (DEGs) were selected based on adjusted p values for False Discovery Rate control (p < 0.01) and fold changes > 2.0. K-mean clustering analysis was performed on DEGs to identify sub-clusters with gene expression patterns activated or inhibited by Nutlin-3a treatment. These activated and inhibited genes were investigated for enriched pathways and gene interaction networks using STRING gene/protein interact ion database [30] and the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8) [31,32]. To prioritize critical genes in Nutlin-3a treatment, the information of gene expression, gene interaction, and enriched pathway functions were integrated using our own designed R software package Rcircle [33].
Statistics
T-test results were calculated using Graphpad Prism 7.05. A two-tailed unpaired non- parametric t-test was computed with the Holm-Sidak method for multiple comparisons with an alpha of 0.05 for the flow cytometry and qRT-PCR data. #p<0.05 and *p<0.01 were considered statistically significant.
Results
P53 expression and localization changes during cardiac differentiation of human induced pluripotent stem cells
To determine if there are significant changes in P53’s temporal dynamics during normal heart development, we utilized time-course microarray data from our previously published transcriptional roadmap of mouse cardiogenesis [33]. Briefly, RNA was extracted from mouse embryonic stem cells (R1), whole hearts at E7.5 and E8.5, and left and right ventricles were collected once they were distinguishable starting at E9.5, D12.5, E14.5, E18.5, 3 days after birth, and continuing out to the adult heart. Time-course gene expression analysis at each time point and ventricle (as appropriate) was conducted as we investigated the temporal expression of P53 and its other two family member genes p63 and p73. The expression heatmap in Supplementary Figure 1 showed that P53 levels were highest in the mouse embryonic stem cells and decreased rapidly after E8.5 and E9.5 with very little expression in the adult mouse heart. This dynamic expression profile suggested that P53 levels could be involved in early regulation of cardiogenesis.
To characterize P53 expression in human cardiogenesis, healthy human induced pluripotent stem cells were differentiated using chemically defined medium (CDM3) with a controlled in vitro differentiation protocol (13). Figure 1A illustrates the timeline for the cardiac differentiation with cells collected for RNA and protein at the indicated days prior to media change. To prove that our cells were moving through a predictable cardiac developmental timeline, we utilized qRT-PCR to look at temporal expression of key markers in cardiac development (Figure 1B). OCT4, SOX2, and NANOG were used to characterize the human iPSCs, T (Brachyury) and GSC expression are mesoderm markers that peaked at Days 1 and 3. Expression of cardiac progenitor genes NKX2-5 and CXCR4 were induced at Day 5, andexpression of cardiomyocyte structural genes MYH7, MYL7, and TNNI3 came on around Day 7 prior to spontaneous beating being observed at Day 8. We also profiled the expression of some of P53’s regulators ATM and ATR in this time course along with key P53 target genes PUMA, NOXA, TSP1, and P21. The levels of ATM and ATR increased intermittently later in the differentiation, around Day 13. Expression levels of PUMA, NOXA, and P21 were all high prior to Day 7, with a resurgence of P21 expression in Day 20 cells. These results indicate that our cells were moving through predictable stages of cardiac development and suggested that P53 dependent processes were changing in a synchronized pattern.
The RNA and protein levels of P53 during in vitro cardiac differentiation time course were assayed using qRT-PCR and Western blot respectively on multiple healthy human iPSC- derived products. Figure 1C shows that P53 RNA levels, normalized to Day 0 hiPSCs, decrease steadily as the differentiated lineages progressed through cardiogenesis. Figure 1D displays a representative blot of P53 protein levels with quantification of three different lines below. P53 protein levels followed a similar pattern predicted by the RNA expression with a slight increase around Day 20. We concluded that P53 levels naturally decline during cardiac differentiation which was consistent with the profile noted in the murine hearts (Supplementary Figure 1).
Since P53’s function as a transcription factor is highly influenced by sub-cellular localization, we utilized immunofluorescence to identify where P53 localized during different stages of cardiac differentiation. We stained for P53 and other relevant markers in Figure 1E and Supplementary Figure 2. In Tra-1-60+ hiPSC colonies we observed mainly cytoplasmic location of P53, whereas induction of differentiation was immediately accompanied with P53 nuclear localization (Day 1). Interestingly, we also observed P53 nuclear localization in NKX2-5 positive cardiac progenitors on Day 5. This suggests P53 is transcriptionally active in these cardiac progenitors. As the cells continue to mature into cardiomyocytes, the P53 localization pattern becomes more diffuse across both the nucleus and cytoplasm with apparent reduction of cellular protein.
Together, these results indicate that P53 is active in different capacities depending on the cell stages during cardiac differentiation. P53 in hiPSCs is known to maintain a strong cytoplasmic presence by priming Bax at mitochondria for rapid apoptosis when P53 is activated [18,19]. P53’s role in cardiac progenitors; however, is less clear, as it has been noted that interactions between P53 and NKX2-5 occurred at a biochemical level in the C2C12 cell line [34].
Post-cardiac specification lineages lose sensitivity to elevated P53 levels
With a baseline for P53 expression during in vitro cardiac differentiation, we investigated the detrimental effects of modulating P53 activity outside of the normal range. We focused on the first ten days of our differentiation protocol which predictably yielded beating cardiomyocytes in this time frame. We utilized a small molecule, Nutlin-3a, as a specific Mdm2 inhibitor that blocks Mdm2 from binding to P53 and thus prevents P53 ubiquitination necessary for degradation. The net result is the accumulation of P53 protein levels in the cell allowing the cells to experience a P53-dependent stress response. Figure 2A outlines the treatment and harvest strategy of a single 24-hour treatment window starting at the stage of iPSC monolayer (Day 0) and continuing out to the Day 10 treatment window. Each treatment window of differentiation was exposed to a dose curve of Nutlin-3a concentrations (5-15 μM) for flow cytometry analysis or a single concentration (10 μM) for RNA and protein. After 24 hours of Mdm2 inhibition, RNA and protein were collected for qPCR analysis and Western blot. Prior to harvesting cells for flow, the iPSC-derived lineages were incubated with EdU and stained with a viability dye to assess cell cycle and cell death, respectively. As seen previously, P53 protein levels steadily decreased as the cells differentiated into cardiomyocytes (Figure 2B). However, the addition of Nutlin-3a reversed this pattern and P53 protein levels accumulated, with the most after Day 4.
However, the early stages prior to Day 4 of differentiation showed less of an increase in P53 levels. We subsequently concluded that this was due to a significant amount of cell death occurring in the iPSC-derived lineages prior to Day 4 (Figure 2C). We detected high levels of cleaved PARP in the early stages of differentiation with Nutlin-3a treatment further confirming a robust apoptosis response prior to Day 4 of differentiation (Figure 2B). Notably, cells past Day 4 were surprisingly resilient to these high P53 levels and showed no viability effects from Nutlin- 3a treatment despite significantly elevated P53 levels. EdU levels overall decreased as cells became cardiomyocytes which is consistent with the reduction of cellular proliferation associated with time of differentiation. Nutlin-3a did cause a decrease of cells in S-phase at all stages of the differentiation (Figure 2D).
We also confirmed the results of these experiments by applying an Mdm4/X inhibitor (SJ172550) to ensure these phenotypes were specific to Mdm2 and P53. We saw no differences in viability or P53 levels with the Mdm4 inhibitor which demonstrated the essential role of P53’s degradation via Mdm2 (data not shown).
To determine the subsequent or latent effects of elevated P53 levels during in vitro cardiac differentiation, we sought to monitor the yield of cardiac lineages that had been previously exposed to a P53-dependent stress test. To accomplish this, we treated iPSC-derived lineages in the same layout as Figure 2A but instead of harvesting after 24 hours, we removed the drug and let the cells continue through differentiation. Figure 2E indicates the days in which cells were treated with Nutlin-3a and the analysis of Day 20 cardiac lineages with cardiac markers cTnI and cTnT. As expected, treatment with Nutlin-3a early in the differentiation caused a dose-dependent decrease in the proportion of cardiomyocytes at Day 20. However, the addition of Nutlin-3a after Day 4 seemed to have the reverse effect by possibly increasing the proportion of cardiomyocytes.
We utilized a pan-caspase inhibitor ZVADFMK to determine if we could rescue the P53- dependent cell death observed prior to Day 4 of differentiation (Supplementary Figure 3). The addition of the caspase inhibitor completely abolished the presence of cleaved PARP (Supplementary Figure 3A). We also conducted flow cytometry with a viability dye to confirm that addition of ZVADFMK reduced the cell death seen with Nutlin-3a treatment (Supplementary Figure 3B). This confirmed that the early iPSC-derived cardiac lineages were being lost through a caspase-dependent apoptotic mechanism.
Upon observing these changes in apoptotic susceptibility, we investigated whether this decrease in sensitivity to elevated p53 levels at later stages of differentiation was due to changes in pro-apoptotic or anti-apoptotic proteins. We measured levels of pro-apoptotic proteins Puma and Noxa, transcriptional targets of p53, and anti-apoptotic proteins Bcl-xL, Mcl1, and Bcl2 (Supplementary Figure 4). Surprisingly, we did not see any notable changes in the anti-apoptotic proteins with Nutlin3a treatment at the RNA or protein level at any day of the cardiac differentiation. Interestingly, Noxa protein levels were high at early stages of cardiac differentiation and showed little response to Nutlin-3a treatment, whereas Puma protein levels were higher at later stages of cardiac differentiation and increased with Nutlin3a treatment.
Additionally, increased expression of p53 downstream target genes such as Mdm2, p21, Puma, Noxa, P21, and Fas was observed past Day 4 of the cardiac differentiation (Supplementary Figure 4B). However, we hypothesized that this lack of increased gene expression in the earlier stages could be due to the increased cell death in those populations. We then concluded that the lack of cell death in the later stages of cardiac differentiation could not solely be attributed to differences in pro- versus anti-apoptotic proteins.
In conclusion, the differential response in iPSC-derived lineages resulting from the transient exposure to P53-stress indicates a shift in P53 sensitivity to P53 resilience within the first week of cardiac specification. Early stages of cardiac differentiation show apoptosis and cell cycle arrest that result in a lack of cardiomyocytes with Nutlin-3a treatment. Surprisingly, cardiac progenitors and cardiomyocytes show a lack of cell death and some decreases in proliferation with Nutlin-3a treatment, but this did not seem to have much effect on the cardiomyocyte output. We also note that this decrease in response to elevated p53 levels around Day 5 of differentiation coincided with reduced cellular proliferation overall. The results of the Nutlin-3a treatments (Figure 2E) indicate that P53 may play different roles in the early stages of cardiac differentiation versus later stages.
Time course transcriptomic profiling of Nutlin-3a treatment revealed transcriptional changes in cellular oxidant detoxification and cell division
To investigate the transcriptome profiles affected by Nutlin-3a treatment, we designed a time course RNA-Seq experiment. It included four critical time points during hiPSC cardiac differentiation in both control and drug-treated groups: Day 3 for cardiac mesoderm stage, Day 5 for cardiac progenitor stage, Day 7 for early cardiomyocytes just before beating stage, and Day 10 for beating cardiomyocyte stage (Figure 3A). These time points allowed us to examine temporal changes in response to Nutlin-3a treatment and better inform the effects p53 was having on the transcriptome. Each group in each time point had 8 samples except for Day 10 which had 6 samples. Samples were treated with DMSO or 10μM Nutlin-3a for 24 hours, and then harvested at the time points indicated to extract mRNA for sequencing. The RNA-Seq files were analyzed with the Map-Rseq [29] pipeline to generate the normalized RPKM gene expression matrix. Principal component analysis (PCA) was performed on the entire transcriptomic RPKM values on all samples in all-time points from both groups. The results revealed a dramatic difference between the control and drug-treated groups (Figure 3B). Then, differentially expressed genes (DEGs) between the treated and control groups were identified with a Generalized Linear Model (GLM). In total, ~1773 DEGs were identified. Unsupervised hierarchical clustering analysis revealed genes with different expression patterns (Figure 3C-1). The following supervised K-mean clustering analysis was performed on these DEGs to cluster them into different sub-clusters based on their expression patterns, with a focus on activated and inhibited gene clusters resulting from Nutlin-3a treatment (Figure 3C-2). Each gene cluster underwent gene interaction network analysis and functional enrichment analysis to investigate their roles in Nutlin-3a treatment during the process of hiPSC cardiac differentiation (Figure 3C-3). In the end, gene expression information, interaction networks, and enriched functions were integrated to highlight important genes (such as hub genes) (Figure 3C-4). The integration results are represented by circle plots where activated and inhibited transcripts are depicted in Figures 4 and 5 respectively.
The sub-cluster of genes activated by Nutlin-3a treatment contained 133 genes, and these were ordered clockwise in the circle plot according to their hierarchical clustering rank (Figure 4). The gene interaction network was mapped to the circular graph with curved lines indicating gene interactions, and wider lines for higher interaction confidence scores from the Gene/Protein Interaction Database (StringDB) [30]. The size of the bubble represents the number of interactions connected to each gene. Genes with larger bubbles were hub genes in the interaction network (Figure 4A).
Enriched function pathway analysis highlighted the top 5 enriched functions (Figure 4B), including cellular oxidant detoxification, regulation of apoptotic processes, response to toxic substances, negative regulation of protein kinase activity, and DNA damage response signal transduction by P53 class mediator resulting in cell cycle arrest. These functions were induced by Nutlin-3a treatment at all stages of differentiation (Figure 4B). Time-course gene expression profiles from the RNA-seq data are also visualized in the circle plot. To save space, the average expression level of the 8 samples in each group was plotted. All the genes in the treated group in each time point had a higher expression level (dark or red-ish) compared with the control group (green-ish) (Figure 4C). Hub genes are indicated in red and included ACTA2, PODNL1, LRRC4C, LRRC66, ANKRD18B, PLK2, DUSP14, DCLK1, RAP2B, CHADL, PRKX, ZNF79, and ZNF561. These genes have more than 10 interaction partners (Figure 4D), and disturbing these genes will have a higher probability of affecting much broader networks and biological functions.
Similar to the layout in the activated cluster as shown in Figure 4, Figure 5 illustrates the sub-cluster of 165 genes inhibited by Nutlin-3a. They are ordered clockwise according to their hierarchical clustering rank, with the interaction network mapped in the middle (Figure 5A), and enriched function pathways in gray circular bands with rectangular color blocks representing genes in those pathways (Figure 6B). Then, average expression levels in each group were plotted in the outer heatmap (Figure 6C). Hub genes with large bubbles and red symbols (Figure 6D) in this sub-group are clustered tightly: KIF2C, BUB1, CDCA8, BUB1B, KIF11, CDK1, CENPA, PLK1, CCNB1, and NDC80. Their functions, closely related to the top 5 enriched function pathways, are involved in cell division, mitotic nuclear division, chromosome segregation, sister chromatid cohesion and mitotic cytokinesis.
Thus, the RNA-Seq results reflected our previous findings in Figure 2 with increased expression of apoptotic and DNA damage response genes and decreased expression of cell division genes after Nutlin-3a exposure during cardiac differentiation of hiPSCs.
Potentialrole for Sestrins and P53 in ROS signaling during cardiomyocyte metabolic maturation
One of the top activated gene functions resulting from Nutlin-3a treatment was cellular oxidant detoxification consisting of the following genes: SESN1, SESN2, P53, INP1, and TXNRD1. SESN1 and SESN2 (Sestrins) are known P53 target genes that help regenerate peroxiredoxins in response to increased reactive oxygen or nitrogen species within cells [35].
Sestrins then provide an important bridge between ROS signaling and P53 activation by acting as antioxidants. Sestrins also mediate the inactivation of mTOR and negatively regulate cell growth when P53 levels are high [36], since mTOR activity is normally inhibited via DNA damage or hypoxia [37].
The process of cardiac differentiation is metabolically quite stressful because glycolysis- utilizing iPSCs must switch to the more efficient mitochondrial oxidative metabolism to meet the high energy needs of beating cardiomyocytes [38]. However, this switch in metabolism invariably leads to increased ROS production, which can be detrimental to cell health if ROS accumulates past a certain level. Previous work linked elevated post-natal ROS levels to the induction of cardiomyocyte cell cycle arrest due to DNA damage signaling [39]. We observed the top RNA-Seq pathways with Nutlin-3a treatment involved up-regulation of cellular oxidant detoxification and down-regulation of cell division. We showed previously that cell proliferation goes down as the cells commit to the cardiac lineage and that Nutlin-3a addition accelerates this process (Figure 2). We utilized qRT-PCR to confirm the increased expression of SESN1 and SESN2 with Nutlin-3a treatment in our RNA-Seq samples (Figure 6).
Thus, Nutlin-3a addition increased P53 levels causing apoptosis in the early stages of cardiogenesis but later stages lost this sensitivity. Our RNA-Seq analysis revealed that increased P53 levels up-regulated genes responsible for cellular oxidant detoxification and apoptosis.
Sestrins are scavengers of ROS and RNS within cells known to be transcriptional targets of P53. Altogether, these results hint at a previously unappreciated role of P53 in the metabolism of cardiac differentiation and confirm that the timing of P53 activation during cardiogenesis revealed disparate responses depending on developmental-stage cell type.
Discussion and Conclusions
Our studies aimed to characterize P53’s expression during cardiac differentiation of hiPSCs and investigate how increasing P53 via small molecular inhibitor Nutlin-3a affected cardiogenesis. Previous microarray data from our lab allowed us to visualize P53 expression during mouse cardiogenesis noting high expression of P53 in mESCs up to E9.5 which then decreased out into the adult heart. We noted a similar pattern in our hiPSC cardiac differentiation with high P53 in hiPSCs and a notable decrease by Day 5 (cardiac progenitor stage). Additionally, in assessing P53’s localization, we noted the high cytoplasmic presence of P53 in the hiPSC colonies and an unexpected shift into the nucleus on Day 5. The presence of P53 in the nucleus of Nkx2-5+ cells to our knowledge has not been observed, and further analysis in these cells via ChIP-Seq would reveal novel insights into P53’s role in cardiac progenitors.
Nkx2-5 and P53 have been shown to interact biochemically in an in vitro system, but remains to be demonstrated in cardiomyocytes specifically [34].
We used small-molecule inhibitor Nutlin-3a to inhibit Mdm2 resulting in P53 protein accumulation during our cardiac differentiation protocol. The use of a small molecule allowed us to treat and harvest in 24-hour time points that would not have been feasible with genetic manipulations. In future studies, we plan to utilize pifthrin-α, a P53 inhibitor, in a comparable manner to these experiments to understand how the loss of P53 in this system would affect cardiogenesis. It should also be noted that we used a minimal differentiation protocol (CDM3) [28] to reduce variability between differentiations and we cannot discount that these results may differ if using another differentiation method.
Nutlin-3a treatments revealed that early stages of cardiac differentiation were quite sensitive to P53 levels with increased caspase-dependent cell death. However, treatment past Day 5 of the cardiac differentiation showed little cell death despite significantly increased protein levels of P53. All stages of the cardiac differentiation showed a G1/S cell cycle arrest following Nutlin-3a treatment. We investigated the expression of pro- and anti-apoptotic proteins after treatment and found that the resistance of the later stages of differentiation to high P53 levels was not due to any obvious changes in the apoptotic machinery
To gain further insights into this differential response to P53, we utilized RNA-Seq to identify transcriptional changes occurring after Nutlin-3a treatment in the mesodermal stage, cardiac progenitor stage, pre-beating immature CMs, and beating CMs. We found the following gene functions and pathways upregulated by Nutlin-3a treatment: cellular oxidant detoxification, apoptosis, toxic substance response, and DNA damage response signal transduction by P53 class mediator resulting in cell cycle arrest. Conversely, our RNA-seq results showed the following important genes were inhibited: KIF2C, BUB1, CDCA8, BUB1B, KIF11, CDK1, CENPA, PLK1, CCNB1, and NDC80. The functions of these inhibited genes were involved in cell division, mitotic nuclear division, chromosome segregation, sister chromatid cohesion and mitotic cytokinesis. We focused on the cellular oxidant detoxification genes upregulated by Nutlin-3a treatment since ROS and RNS scavenging is considered a non-canonical function of P53. Of these genes, the Sestrins (SESN1 and SESN2) were established P53 target genes stimulated in response to ROS. Though well-known as a tumor suppressor, P53 orchestrates cellular responses to various kinds of molecular signals. One of these signals is reactive oxygen species (ROS) which acts as both an upstream signal to activate P53 and a downstream signal that regulates apoptosis. The crosstalk between p53 and the ROS signaling pathway is critical for cell differentiation and normal organ development [40] Our study illustrates that p53 may be working with the ROS signaling pathway via Sestrins (SESN1 and SESN2) during cardiac differentiation of hiPSCs. These results shed light on a previously unappreciated role of P53 in the metabolism of cardiac differentiation.
Previous work has already illustrated the critical role for the P53/Mdm2 signaling axis in murine adult heart physiology and metabolism. A conditional knock-out of P53 in the adult murine heart caused spontaneous hypertrophy and RNA-Seq data revealed that loss of p53 negatively impacted Complex I and III activity, and fatty acid oxidation and glucose metabolism were de-regulated [41]. Similarly to the embryo, loss of Mdm2 in the adult murine heart lead to cardiac hypertrophy and subsequent dysfunction after only a few days. As in our study, the authors observed cardiomyocyte cell death, inhibition of antioxidant systems, and impaired mitochondrial bioenergetics [42].
One caveat of our in vitro system is that the cells are grown two-dimensionally in normoxia which invariably affects their metabolism. However, it was encouraging to see that our results reflected those seen by Hauck, et al [42]. The metabolic state of iPSCs differentiating into cardiomyocytes changes drastically in a short period of time and depends critically on proper mitochondrial function [38]. Since one of the top hits in our RNA-Seq from inhibiting Mdm2 function was cellular oxidant detoxification, we hypothesize that P53 may play a critical role in ROS handling during cardiac differentiation. The role of the Sestrins in cardiomyocytes has been little explored but is known to ameliorate cardiac hypertrophy through the mTOR pathway [43,44] or to respond to cardiotoxic agents such as doxorubicin [45]. We believe that the interactions between the cell stress response pathways activated by P53 and the metabolism changes required for cardiac differentiation could show further interactions. Thus, it is critical to gain a better understanding of the roles P53 is playing in cardiogenesis as its influence can be felt not just in cell survival and proliferation, but in metabolism, autophagy, mitochondrial stability, and more. Understanding these intersections between environmental changes and cell responses in this human cardiac differentiation system will better inform the pathogenesis of CHDs and provide a more detailed understanding of human heart development.
Acknowledgments
This work was supported by the Todd and Karen Wanek Family Program for Hypoplastic Left Heart Syndrome at Mayo Clinic. We thank ReGen Theranostics for the human induced pluripotent stem cells. Emma Brandt was supported by the Gary and Anita Klesch Predoctoral Fellowship. The Mayo Microscopy and Cell Analysis Core was utilized for the confocal images. The Center for Regenerative Medicine provided access to qRT-PCR, flow cytometry, and western blot resources. Frank Secreto and Katie Minter Dykhouse provided valuable editorial commentary and experimental guidance.
Conflicts of Interest
Mayo Clinic and author Timothy J. Nelson have financial rights to ReGen Theranostics through licensing agreements. All other authors have no conflict of interest.
- Donehower LA (1992) Mice Deficient for P53 Are Developmentally Normal but Susceptible to Spontaneous Tumors. Nature 356: 215-21.
- Sah VP (1995) A subset of p53-deficient embryos exhibit exencephaly. Nature Genetics 10, 175-80.
- de Oca Luna RM, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203-6.
- Jones SN, Roe AE, Donehower LA, BradleyA (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378: 206-8.
- Choi J, Donehower LA (1999) p53 in embryonic development: maintaining a fine balance. Cell Mol Life Sci 55: 38-47.
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296-9.
- Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G (2006) Tissue- Specific Differences of p53 Inhibition by Mdm2 and Mdm4. Molecular and Cellular Biology 26: 192.
- Zhang Q (2012) Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J Pathol 228: 416-28.
- Van Nostrand JL (2014) Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514: 228-32.
- Caprio C, Baldini A (2014) p53 suppression partially rescues the mutant phenotype in mouse models of DiGeorge syndrome. Proceedings of the National Academy of Sciences 111: 13385.
- Morgan SC (2008) Cardiac outflow tract septation failure in Pax3-deficient embryos is due to p53-dependent regulation of migrating cardiac neural crest. Mechanisms of Development 125: 757-67.
- Su D (2017) ROCK1/p53/NOXA signaling mediates cardiomyocyte apoptosis in response to high glucose in vitro and vivo. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1863: 936-46.
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663-76.
- Takahashi K (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861-72.
- Maimets T, Neganova I, Armstrong L, Lako M (2008) Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene 27: 5277-87.
- Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R (2010) p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis 31: 1501-8.
- Yang A (2011) Nucleolin Maintains Embryonic Stem Cell Self-renewal by Suppression of p53 Protein-dependent Pathway. Journal of Biological Chemistry 286: 43370-82.
- Dumitru R (2012) Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol Cell 46: 573-83.
- Setoguchi K, TeSlaa T, Koehler CM, Teitell MA (2016) P53 Regulates Rapid Apoptosis in Human Pluripotent Stem Cells. Journal of Molecular Biology 428: 1465-75.
- Qin H (2007) Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem 282: 5842-52.
- Jain AK (2012) p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol 10: e1001268.
- Wang Q (2017) The p53 Family Coordinates Wnt and Nodal Inputs in Mesendodermal Differentiation of Embryonic Stem Cells. Cell Stem Cell 20: 70-86.
- Hadjal Y, Hadadeh O, Yazidi CE, Barruet E, Binetruy BA (2013) p38MAPK-p53 cascade regulates mesodermal differentiation and neurogenesis of embryonic stem cells. Cell Death Dis 4: e737.
- Moore IV JB (2016) The Epigenetic Regulator HDAC1 Modulates Transcription of a Core Cardiogenic Program in Human Cardiac Mesenchymal Stromal Cells Through a p53-Dependent Mechanism. STEM CELLS 34: 2916-29.
- Vassilev LT ((2004) In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 303: 844.
- Secreto FJ (2017) Quantification of Etoposide Hypersensitivity: A Sensitive, Functional Method for Assessing Pluripotent Stem Cell Quality. Stem cells translational medicine 6: 1829-39.
- Perales-Clemente E (2016) Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. The EMBO Journal 35: 1979-90.
- Burridge PW, Holmström A, Wu JC (2015) Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Curr Protoc Hum Genet 87: 21-3.
- Kalari KR (2014) MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics 15: 224.
- Szklarczyk D (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research 45: D362-8.
- Huang DW, Sherman BT, Lempicki RA (2008) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 37: 1-13.
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4: 44-57.
- Li X (2014) Transcriptional atlas of cardiogenesis maps congenital heart disease interactome. Physiological Genomics 46, 482-95.
- Kojic S (2015) Cardiac transcription factor Nkx2.5 interacts with p53 and modulates its activity. Arch Biochem Biophys 569: 45-53.
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM (2004) Regeneration of Peroxiredoxins by p53-Regulated Sestrins, Homologs of Bacterial AhpD. Sci 304: 596.
- Budanov AV, Karin M (2008) p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 134, 451-60.
- Levine AJ, Feng Z, Mak TW, You H, Jin S (2006) Coordination and communication between the p53 and IGF-1–AKT–TOR signal transduction pathways. Genes & Development 20: 267-75.
- Chung S (2007) Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature Clinical Practice Cardiovascular Medicine 4: S60-7.
- Puente Bao N (2014) The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 157: 565-79.
- Liu B, Chen Y, St Clair DK (2008) ROS and p53: A versatile partnership. Free Radical Biology and Medicine 44: 1529-35.
- Mak TW, Hauck L, Grothe D, Billia F (2017) p53 regulates the cardiac transcriptome. Proc Natl Acad Sci U S A 114, 2331-6.
- Hauck L (2017) Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One 12: e0189861.
- Xue R (2017) Sestrin 1 ameliorates cardiac hypertrophy via autophagy activation. J Cell Mol Med 21: 1193-205.
- Dong B, Xue R, Sun Y, Dong Y, Liu C (2017) Sestrin 2 attenuates neonatal rat cardiomyocyte hypertrophy induced by phenylephrine via inhibiting ERK1/2. Molecular and Cellular Biochemistry 433: 113-23.
- Li R (2019) Cardioprotective roles of sestrin 1 and sestrin 2 against doxorubicin cardiotoxicity. American Journal of Physiology-Heart and Circulatory Physiology 317: H39-48.
FIGURE 1
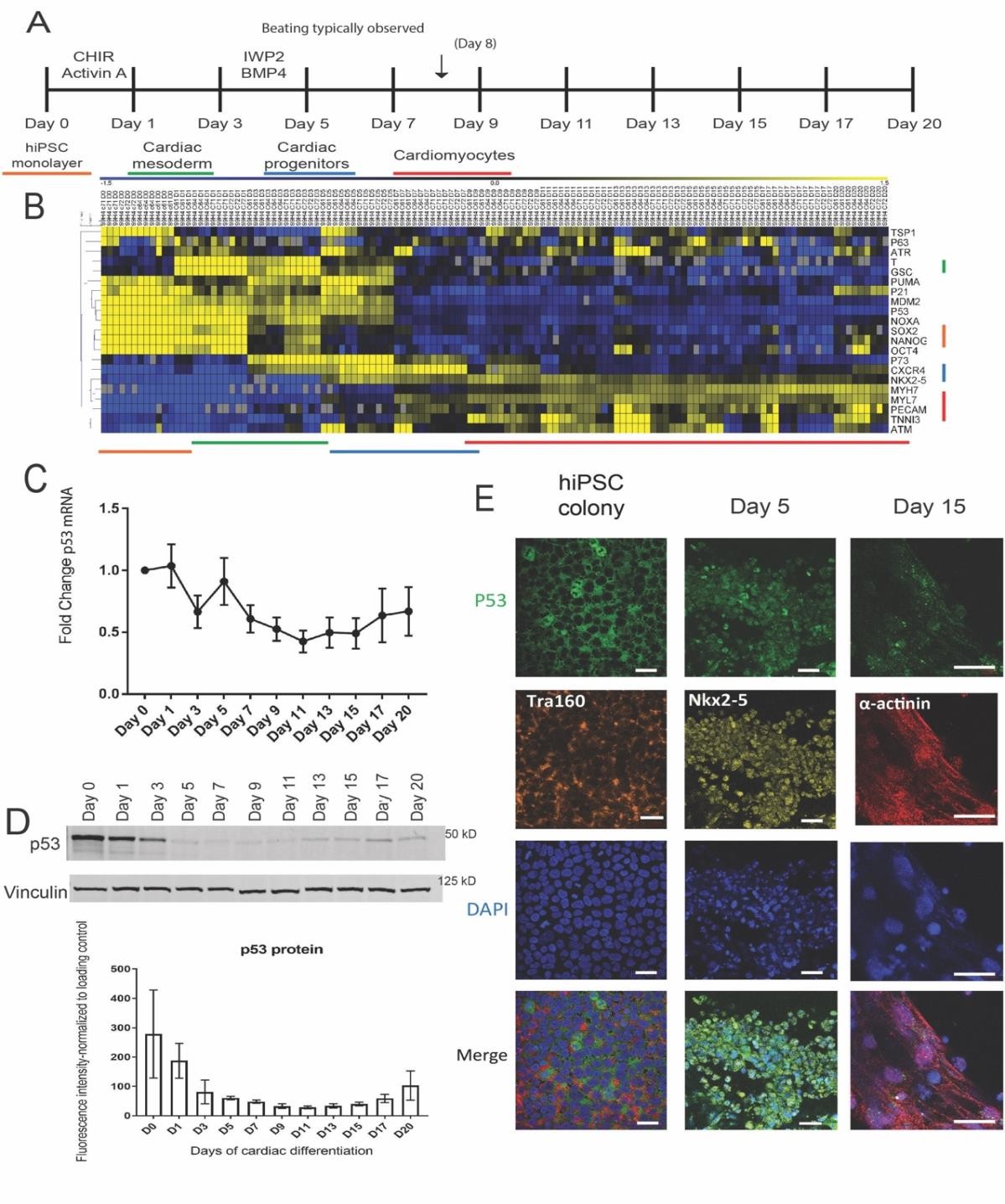
Figure 1: P53 expression and localization changes during cardiac differentiation
(A) Timeline of human in vitro cardiac differentiation of hiPSCs with indicated small molecules used and the days in which samples were collected for RNA, protein, and immunostaining. Developmental stages are indicated by color underneath their corresponding days. (B) RNA was collected from the indicated time points in A and subjected to qRT-PCR and the results compiled into a heatmap. The genes were grouped temporally based on their expression levels with genes identifying pluripotency (orange), cardiac mesoderm (green), cardiac progenitors (blue), and cardiomyocytes (red). These stages are color coded and correlate back to 2A. (n = 4) (C) qRT-PCR of P53 mRNA expression across the cardiac differentiation. (n = 28). (D) Representative western blot and quantification of P53 protein expression during cardiac differentiation (n = 3). (E) Immunostaining for P53 in human iPS colonies (Tra-1-60+), Day 5-cardiac progenitors (NKX2-5 +), and in Day 15 cardiomyocytes (α-actinin +). Scale bars = 20 μm
FIGURE 2
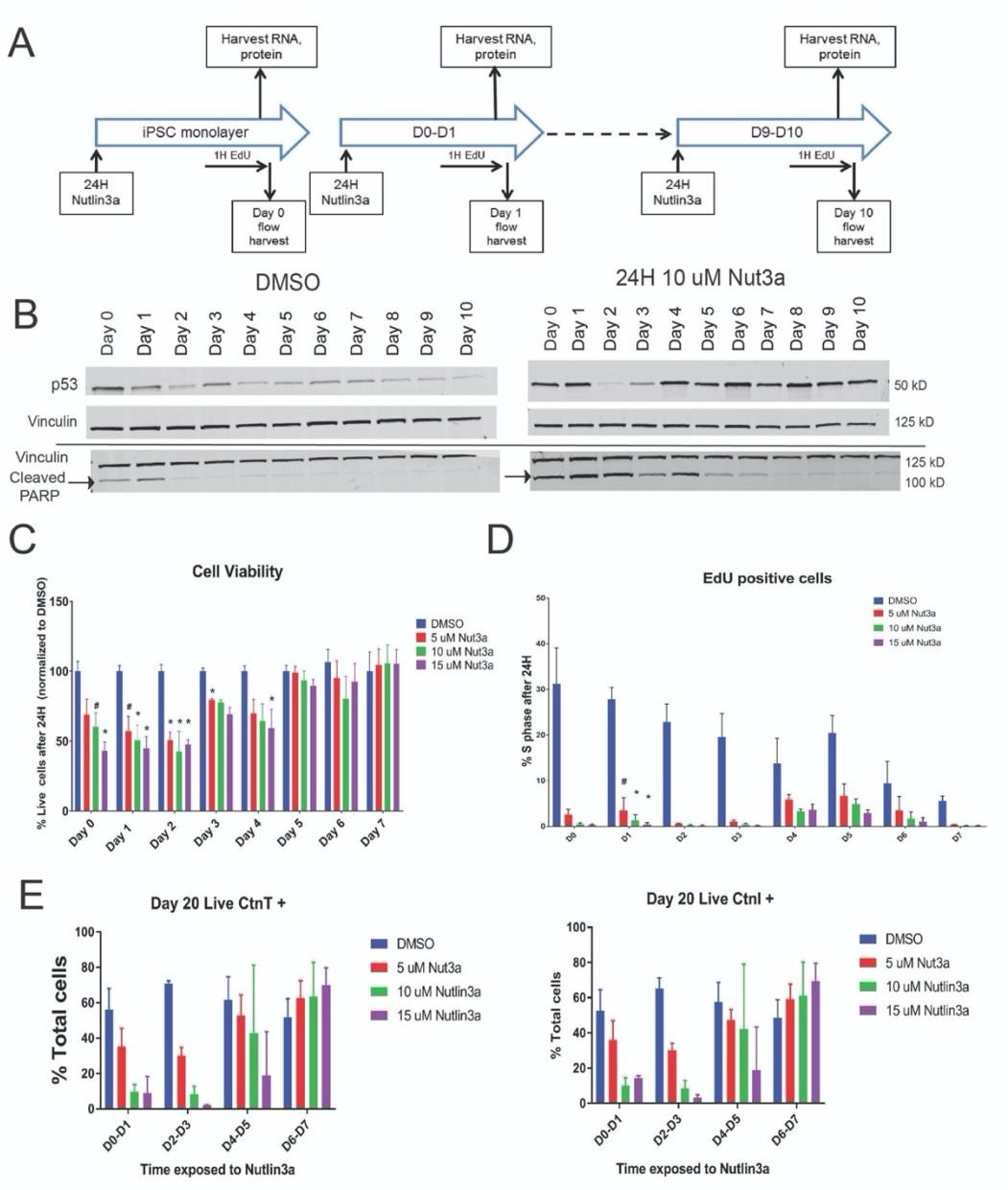
Figure 2: Early stages of cardiac differentiation show increased sensitivity to P53 levels. (A) Time line of sample collection during the first 10 days of cardiac differentiation with or without Nutlin-3a exposure in 24 hour increments. (B) Representative western blot of P53 protein levels and cleaved PARP at each day of the differentiation treated either with DMSO (vehicle)or 10 uM of Nutlin-3a for 24 hours. Vinculin is included as a loading control. The black line indicates the samples were run on separate gels (n=2).
(C) After the 24 hour incubation with Nutlin-3a, each day of differentiation was incubated with EdU for 1 hour, stained with a Live- Dead viability dye and then subjected to flow cytometry to assess cell death and proliferation. An increasing concentration of Nutlin- 3a showed a dose-dependent increase in cell death in cells younger than Day 4. Nutlin-3a treatment had little effect on cell viability for cells older than Day 4 (n ≥ 4 each day). (D) The same population of cells in (C) were stained for EdU and DAPI to identify the number of cells in S phase. There was a dose-dependent decrease in the % of EdU+ cells in S phase with increasing Nutlin-3a concentration with the majority of the cells remaining in G1. The amount of proliferating cells also decreased naturally as the cells continued to differentiate (n ≥ 2 each day). (E) Cells at the time points indicated were exposed to a dose curve of Nutlin-3a for 24 hours, after which the drug was removed, and the cells were cultured as normal out to Day 20. They were then stained with a Live- Dead viability dye and cardiac markers cTnI and cTnT to assess cardiomyocyte output. Addition of Nutlin-3a prior to Day 4 caused a dose-dependent decrease in the proportion of cardiomyocytes produced. Addition of Nutlin-3a past Day 5 slightly increased the proportion of cardiomyocytes (n=2) (#: p< 0.05, *: p< 0.01).
FIGURE 3
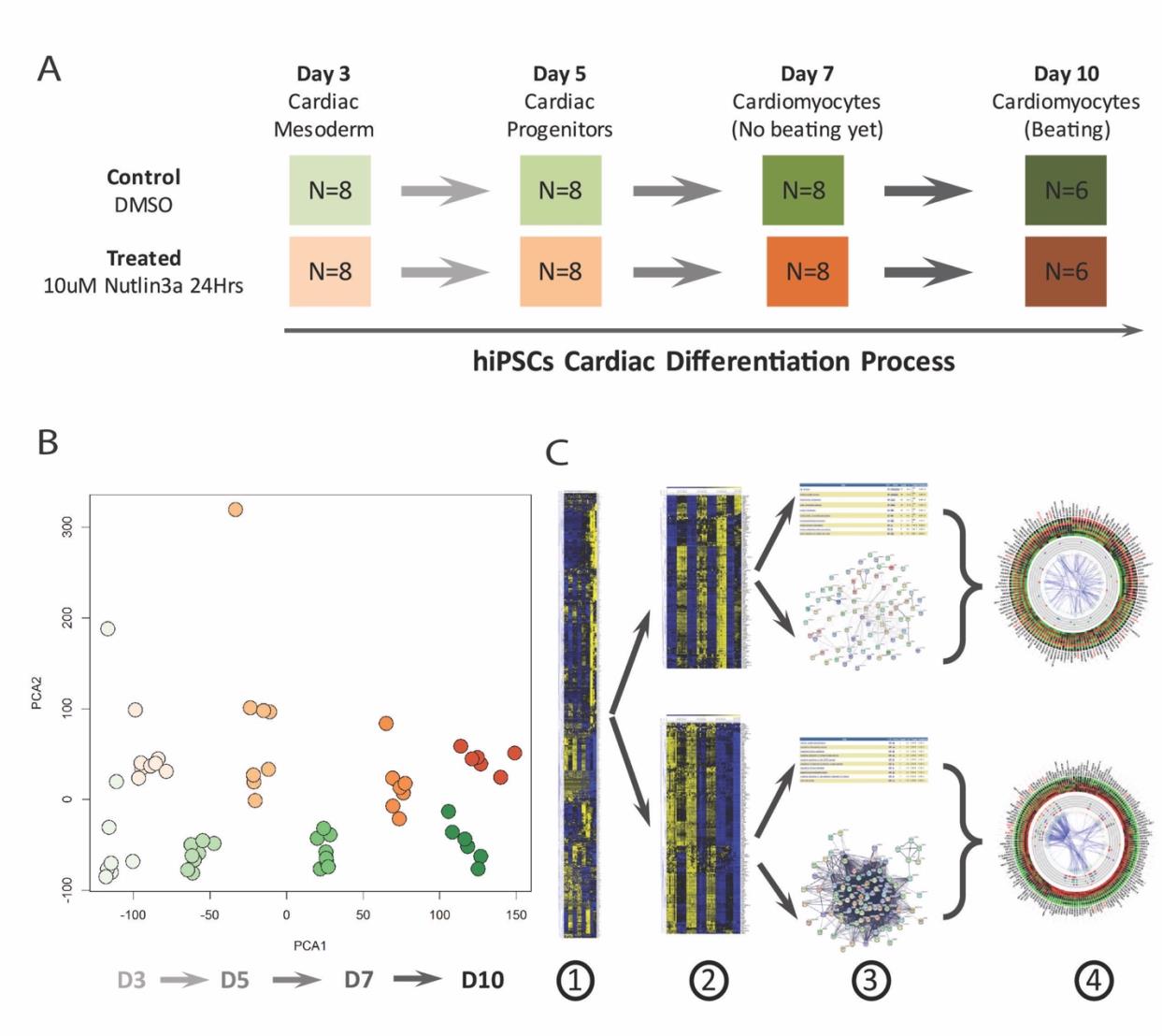
Figure 3: RNA-Seq study revealed different gene expression profiles between the control and Nutlin-3a treated samples during hiPSCs cardiac differentiation
(A) Time course RNA-Seq experimental design included 4 time points in both control and Nutlin-3a treated groups: Day 3 Cardiac Mesoderm, Day 5 Cardiac Progenitors, Day 7 Cardiomyocytes (No beating yet), Day 10 Cardiomyocytes (Beating). Each group in each time point had 8 samples except for Day 10 which had 6 samples. Treated groups were treated with 10uM Nutlin-3a for 24 hours. Samples were collected at each time point to extract mRNA for sequencing. (B) PCA was performed on all samples from both groups (orange: Nutlin-3a, green: control). The results revealed a dramatic difference between control and drug- treated groups. (C) The following differential analysis between both groups at all time points highlighted genes displaying variable patterns of differential expression between the two groups (1). K-mean cluster analysis was performed to cluster the genes into different sub-clusters based on their expression patterns, with focus on activated and inhibited gene clusters shown (2). Each gene cluster underwent gene interaction network analysis and function enrichment analysis to investigate their roles in Nutlin-3a treatment (3). At the end, gene expression information, interaction networks, and enriched functions were integrated together to highlight important genes (such as hub genes) (4). The integration results are shown in the following Figures 4 & 5.
FIGURE 4
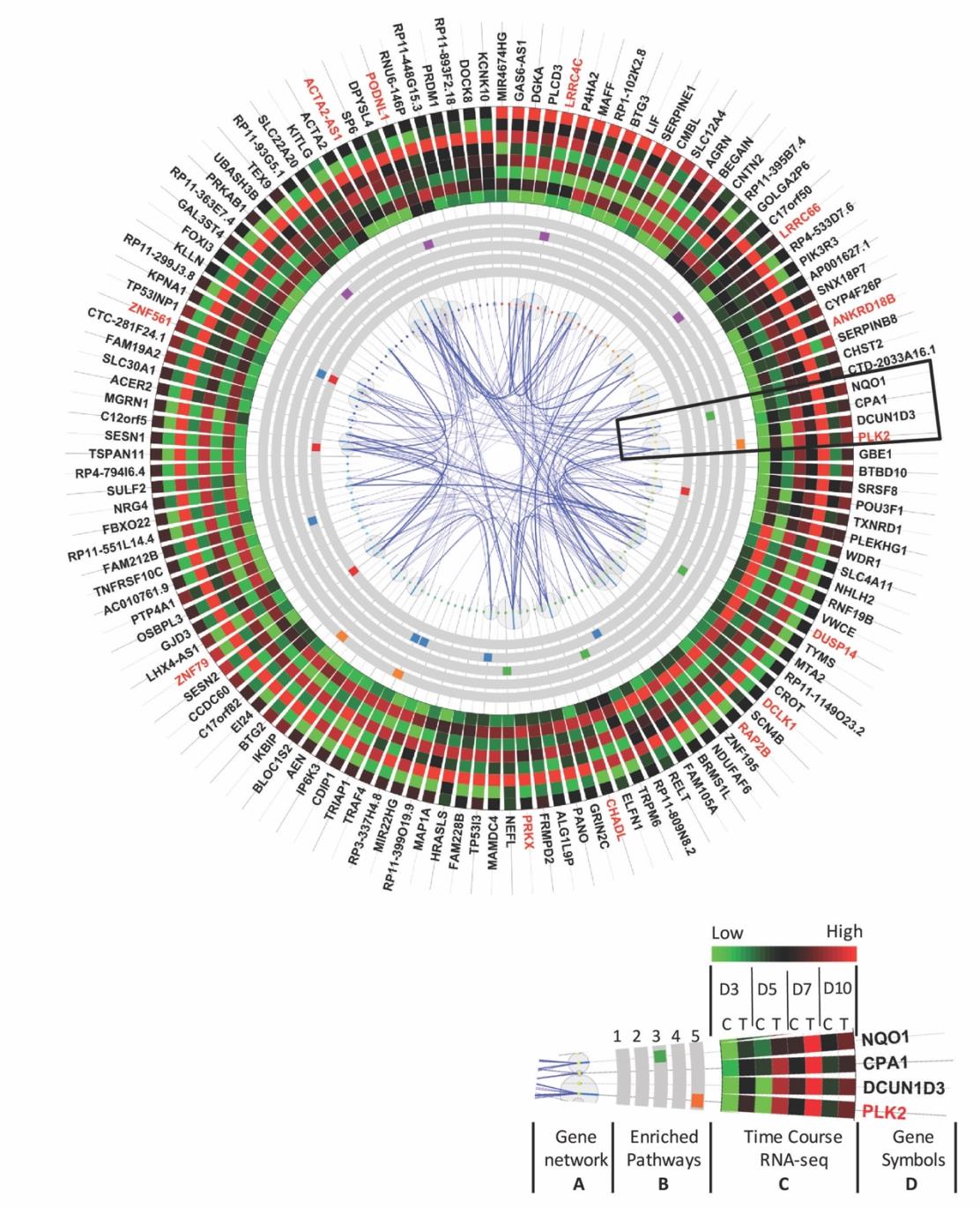
Figure 4: Genes activated by Nutlin-3a treatment highlight gene functions involved in cellular oxidant detoxification, apoptosis, DNA damage response related to P53
(A) Gene interaction network lines indicate the gene interactions with wider lines for higher interaction scores from the Gene/Protein Interaction Database (String DB). The bubble size represents the number of interactions connected to that gene. Genes with larger bubbles are hub genes in the interaction network. (B) Top 5 enriched function pathways for Nutlin-3a activated genes. 1: Cellular oxidant detoxification. 2: Regulation of apoptotic process. 3: Response to toxic substance. 4: Negative regulation of protein kinase activity. 5: DNA damage response, signal transduction by P53 class mediator resulting in cell-cycle arrest. (C) Time course gene expression profiles from RNA-Seq data with green for lower expression and red for higher expression. Time course RNA-Seq design included critical time points: Day 3 (D3), 5, 7, and 10. Each time point contained two groups: control (C) and Nutlin-3a-treated group (T). To save space, average expression level of 8 samples in each group was plotted. (D) Gene symbols with genes with more than 10 interaction partners are labeled in red. These red genes are hubs in the interaction network.
FIGURE 5
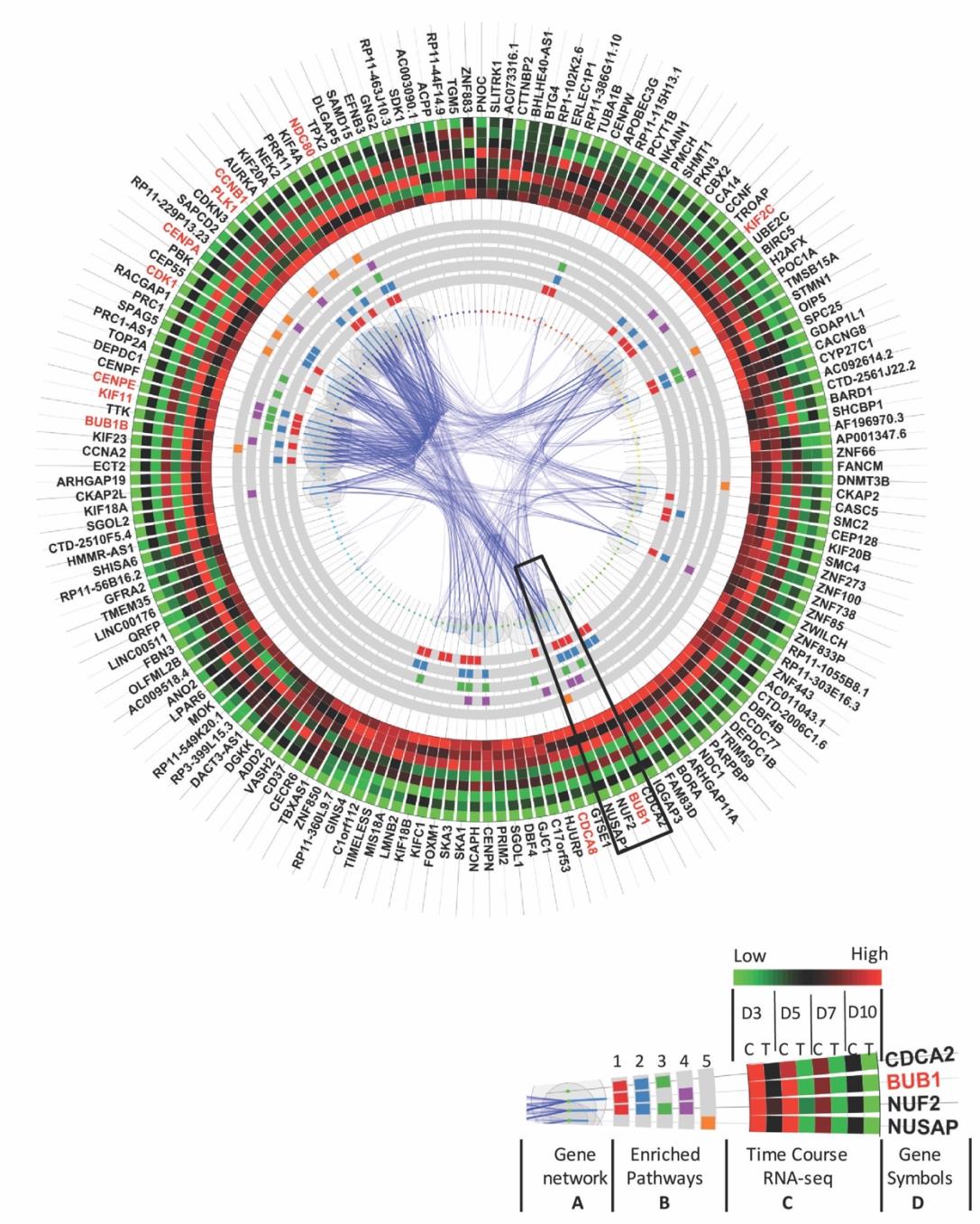
Figure 5: Genes inhibited by Nutlin-3a treatment demonstrated functions relating to cell division, mitotic nuclear division and cytokinesis
(A) Gene interaction network lines indicate the gene interactions with wider lines for higher interaction scores from the Gene/Protein Interaction Database (String DB). The bubble size represents the number of interactions connected to that gene. Genes with larger bubbles are hub genes in the interaction network.
(B) The top 5 enriched function pathways inhibited by Nutlin-3a treatment included 1: Cell division, 2: Mitotic nuclear division, 3: Chromosome segregation, 4: Sister chromatid cohesion, 5: Mitotic cytokinesis. (C) Time course gene expression profiles from RNA-Seq data with green for lower expression and red for higher expression. Time course RNA-Seq design included critical time points: Day 3 (D3), 5, 7, and 10. Each time point contained two groups: control (C) and Nutlin-3a-treated group (T). To save space, average expression level of 8 samples in each group was plotted. (D) Gene symbols with genes with more than 30 interaction partners are labeled in red. These red genes are hubs in the interaction network.
FIGURE 6
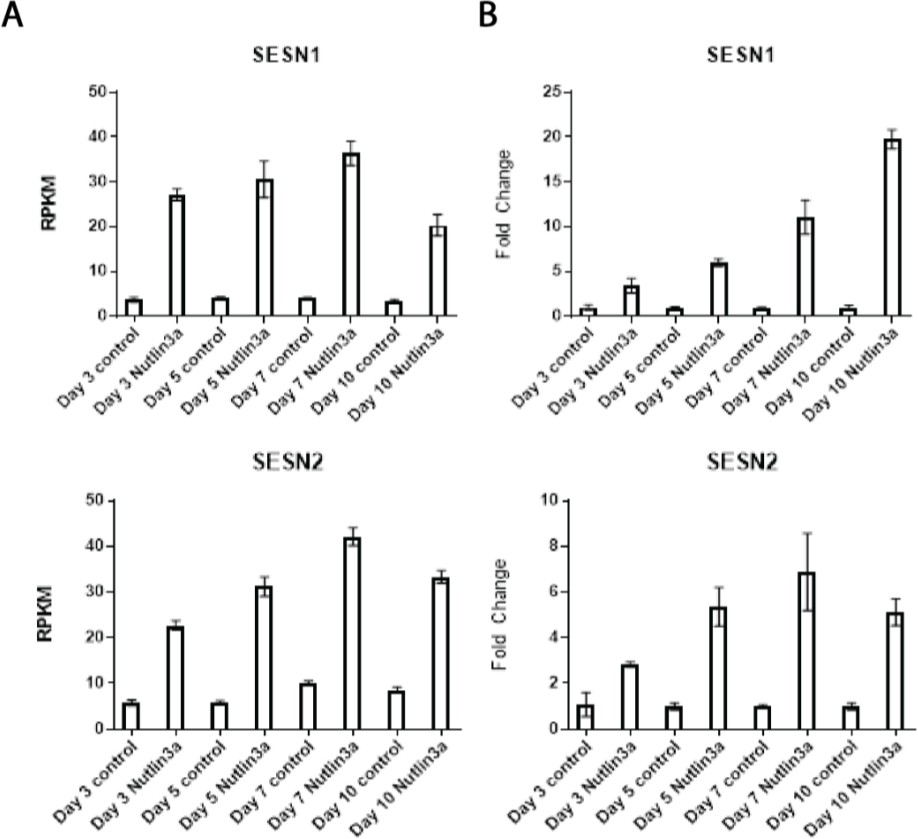
Figure 6: Nutlin-3a causes anti-oxidant Sestrin gene increases in cardiomyocytes. RNA-Seq revealed response to oxidative stress as one of the top pathways activated by Nutlin-3a with Sestrin1 and Sestrin2 implicated. These changes were validated by qRT- PCR on the right (n=3) and the original RPKM values on the left ( n ≥ 6)
FIGURE 7
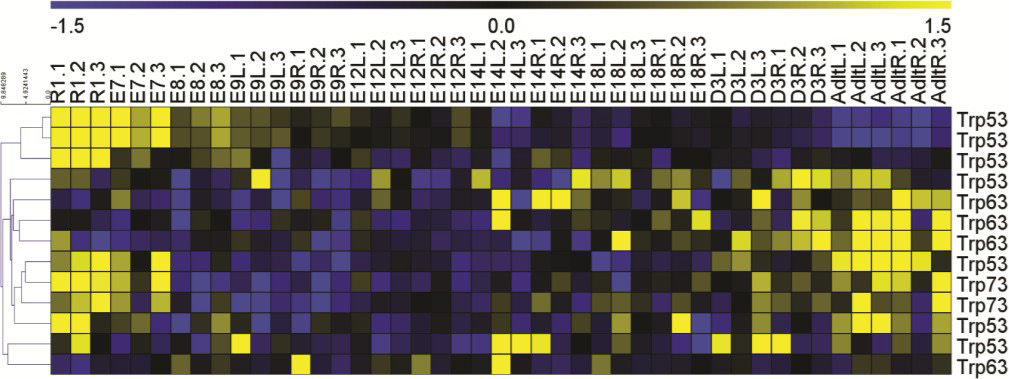
Supplementary Figure 1: Gene expression patterns of P53 family members during murine cardiogenesis. This time-course microarray heatmap showed the progression of expression of P53 family members starting in R1 mouse ESCs that begins at E7.5 and ends with tissue from the adult mouse heart. Samples were collected from both left and right ventricles once they were morphologically distinguishable starting from E9.5 (L: Left Ventricle; R: Right Ventricle). The multiple probes for each gene tended to cluster together in their distinct patterns with only a few statistically insignificant outliers, potentially due to probe sensitivity and cross-hybridization.
FIGURE 8
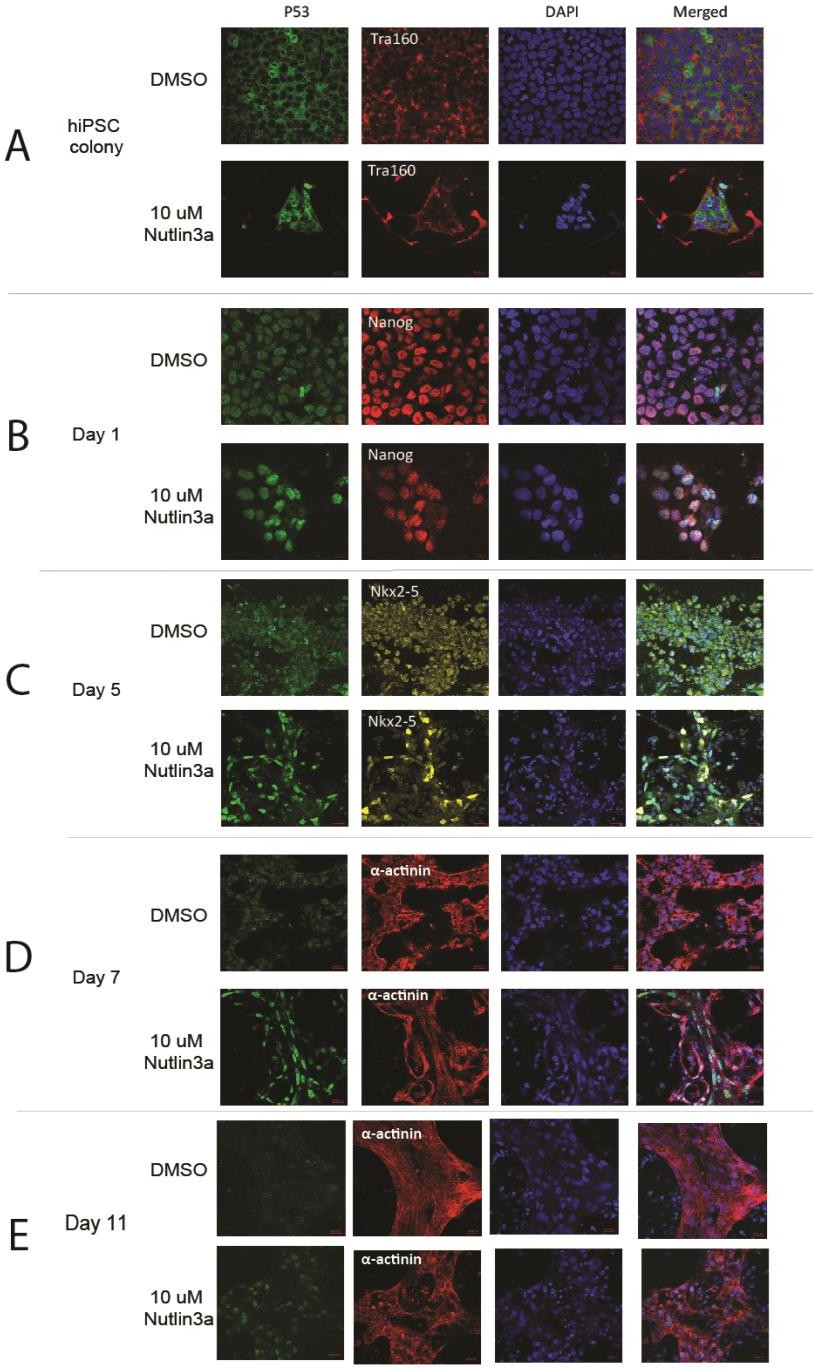
Supplementary Figure 2: Immunofluorescence images of P53 localization during cardiac differentiation with Nutlin-3a treatment
Cells were treated with DMSO or 10 μM Nutlin-3a for 24 hours then fixed and stained for P53 and an appropriate cell stage marker. (A) Human induced pluripotent stem cell colonies. (B) Day 1 Nanog + cells. (C) Day 5 NKX2-5 + cardiac progenitors. (D) Day 7 cardiomyocytes not yet beating.
(E) Day 11 cardiomyocytes that were spontaneously beating. Treatment with Nutlin-3a in all stages except hiPSC colonies resulting in increased nuclear staining of P53.
FIGURE 9
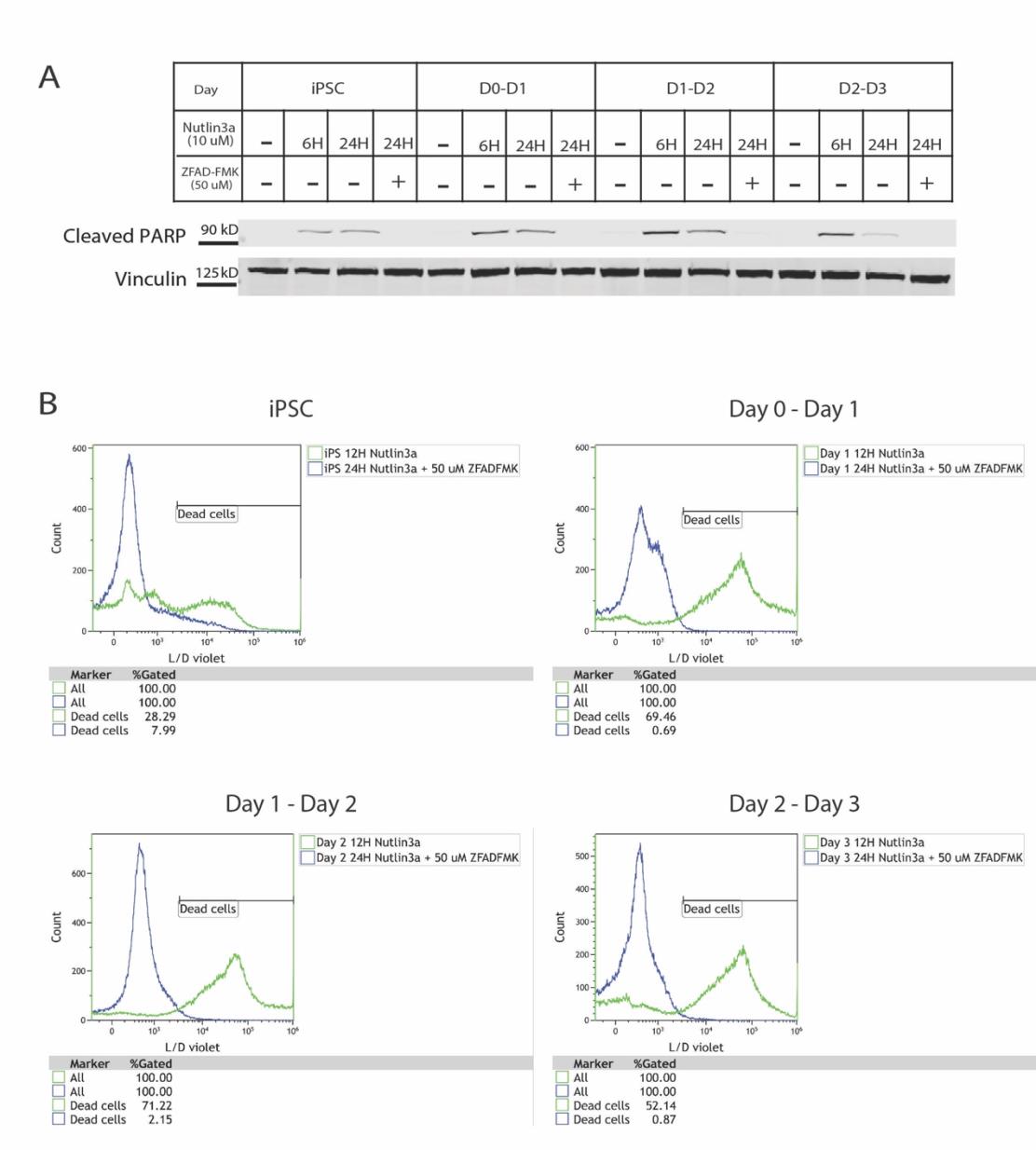
Supplementary Figure 3: IIncreased apoptosis in early stages of cardiac differentiation was prevented by addition of a pan-caspase inhibitor
(A) Each time point of the differentiation was incubated with either 10 μM Nutlin-3a for 6, 12, and 24 hours or 10 μM Nutlin-3a with 50 μM ZFAD-FMK, a pan-caspase inhibitor. A representative western blot of cleaved PARP revealed that ZFAD-FMK incubated alongside Nutlin-3a eliminated the presence of cleaved PARP in the early stages of differentiation. (B) Flow cytometry of cells incubated with 10 μM Nutlin-3a for 12 hours or 10 μM Nutlin-3a for 24 hours plus 50 μM ZFAD-FMK. Cells were stained with a Live-Dead viability dye and the number of dead cells quantified. Addition of ZFAD-FMK caused a dramatic reduction in cell death in these early stages of cardiac differentiation.
FIGURE 10
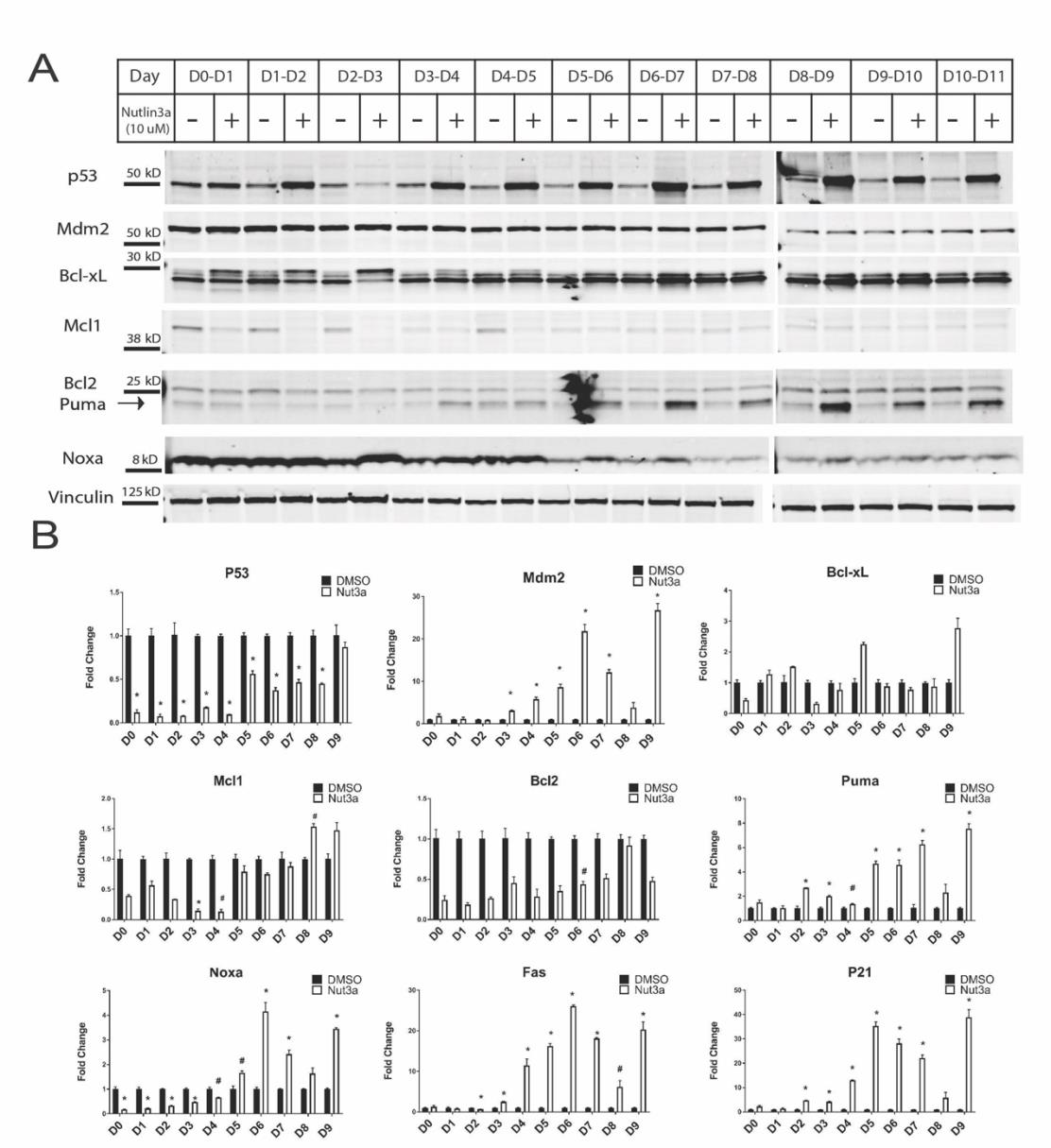
Supplementary Figure 4: Cardiomyocytes do not undergo apoptosis despite increased P53 and Puma levels. (A) Western blot for pro- and anti-apoptotic protein levels each day of cardiac differentiation after 24 hours of Nutlin-3a treatment. Vinculin was included as a loading control. (n=2) (B) qRT-PCR analysis of pro- and anti-apoptotic genes influenced by P53 signaling. (n=3) (#p<0.05, *p<0.01)
Figures at a glance